Luisa Ricciardi, Fabiana Furci, Antonio Ieni, Antonio Macrì
{"title":"Castleman Disease in a Patient with Common Variable Immunodeficiency.","authors":"Luisa Ricciardi, Fabiana Furci, Antonio Ieni, Antonio Macrì","doi":"10.1155/2019/5476383","DOIUrl":null,"url":null,"abstract":"<p><p>Common variable immunodeficiency (CVID) is a primary immunodeficiency due to a disorder of the adaptive immune system which causes hypogammaglobulinemia and therefore an increased susceptibility to infection; noninfectious, inflammatory conditions including systemic autoimmunity and lymphoproliferative complications are also commonly associated with CVID. Castleman disease (CD) is a systemic disease clinically characterized by diffuse lymphadenopathy, splenomegaly, anemia, and systemic inflammatory symptoms. This makes CD a great mimicker of more common benign and malignant masses in the neck, chest, abdomen, and pelvis. A novel case of primary immunodeficiency (CVID) in a middle-aged woman, who developed multicentric CD (MDC) with splenomegaly, is described. The authors suggest that the onset of MCD and of the correlated splenomegaly was due to incorrect management of the hypogammaglobulinemia as immunoglobulin G (IgG) levels were not kept within normal ranges. Correct management of the hypogammaglobulinemia allowed splenectomy to be performed without any infectious surgical complications. MCD is reported for the first time in association with an adult case of CVID. The above reported case highlights the need for a timely correct diagnosis and treatment of CVID to avoid complications, which could cause recourse to splenectomy, such as in our case or development of malignancies.</p>","PeriodicalId":42865,"journal":{"name":"Case Reports in Immunology","volume":" ","pages":"5476383"},"PeriodicalIF":1.5000,"publicationDate":"2019-02-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2019/5476383","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Immunology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2019/5476383","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2019/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
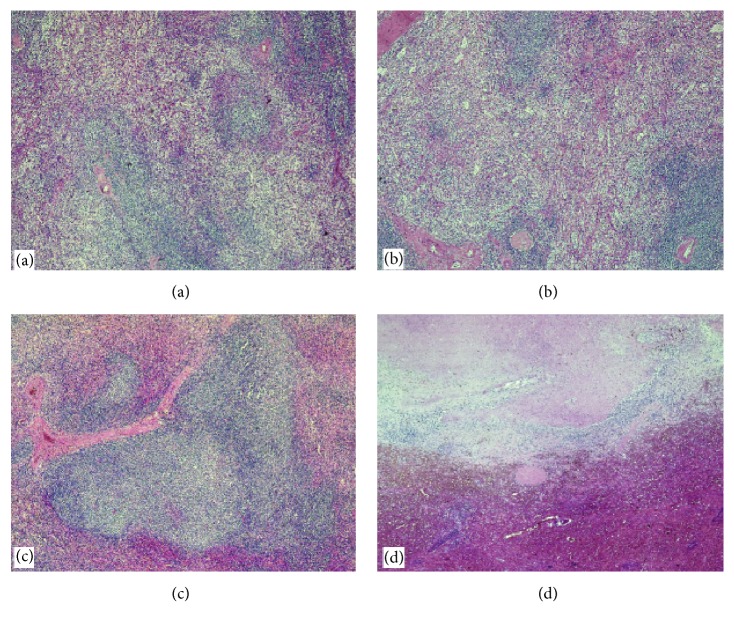
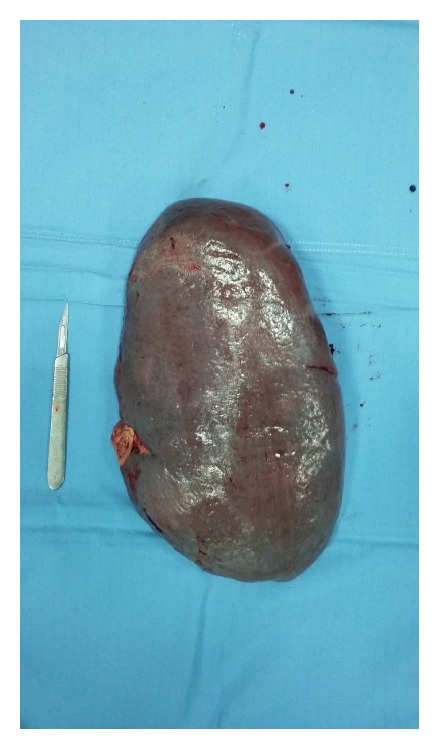
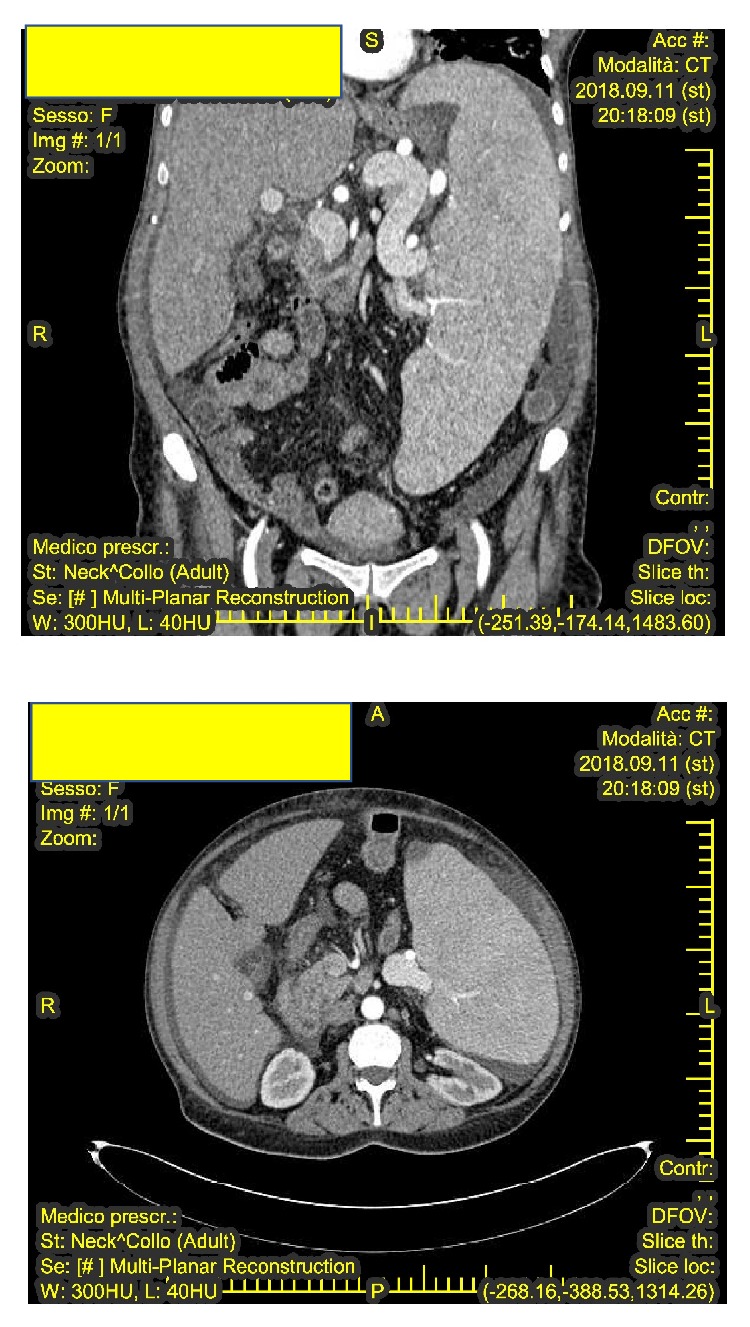
Common variable immunodeficiency (CVID) is a primary immunodeficiency due to a disorder of the adaptive immune system which causes hypogammaglobulinemia and therefore an increased susceptibility to infection; noninfectious, inflammatory conditions including systemic autoimmunity and lymphoproliferative complications are also commonly associated with CVID. Castleman disease (CD) is a systemic disease clinically characterized by diffuse lymphadenopathy, splenomegaly, anemia, and systemic inflammatory symptoms. This makes CD a great mimicker of more common benign and malignant masses in the neck, chest, abdomen, and pelvis. A novel case of primary immunodeficiency (CVID) in a middle-aged woman, who developed multicentric CD (MDC) with splenomegaly, is described. The authors suggest that the onset of MCD and of the correlated splenomegaly was due to incorrect management of the hypogammaglobulinemia as immunoglobulin G (IgG) levels were not kept within normal ranges. Correct management of the hypogammaglobulinemia allowed splenectomy to be performed without any infectious surgical complications. MCD is reported for the first time in association with an adult case of CVID. The above reported case highlights the need for a timely correct diagnosis and treatment of CVID to avoid complications, which could cause recourse to splenectomy, such as in our case or development of malignancies.
期刊介绍:
Case Reports in Immunology is a peer-reviewed, Open Access journal that publishes case reports and case series related to allergies, immunodeficiencies, autoimmune diseases, immune disorders, cancer immunology and transplantation immunology.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
