Oana Neagoe, Anda Ciobanu, Rodica Diaconu, Oana Mirea, Ionuț Donoiu, Constantin Militaru
{"title":"A rare case of familial restrictive cardiomyopathy, with mutations in MYH7 and ABCC9 genes.","authors":"Oana Neagoe, Anda Ciobanu, Rodica Diaconu, Oana Mirea, Ionuț Donoiu, Constantin Militaru","doi":"10.15190/d.2019.12","DOIUrl":null,"url":null,"abstract":"<p><p>Restrictive cardiomyopathy is the least common type of cardiomyopathy, being defined by diastolic dysfunction and often unimpaired systolic function. Restrictive cardiomyopathies can be classified as familial or non-familial. Patients with familial restrictive cardiomyopathy can develop signs and symptoms of this condition anytime from childhood to adulthood. The evolution of the disease is towards signs and symptoms of pulmonary and systemic congestion and, without treatment, there is a five-year mortality rate of approximately 30% in these patients. We discuss the case of a 43-year-old patient diagnosed with familial restrictive cardiomyopathy with positive genetic tests for mutations of MYH7 gene and ABCC9 gene, who was first hospitalized in 2011 for palpitations. The echocardiography performed in evolution showed a continuous alteration of right ventricle function, without important differences of left ventricular function. She developed heart failure symptoms six years after diagnosis and she had seven hospitalizations in the past two years, currently with an increasing need of diuretics and persistent hepatic dysfunction. Cardiac transplantation or left ventricular assist device therapy should be considered in patients with severe heart failure symptoms and no longer effective treatment. However, elevated pulmonary vascular resistance excludes the patient from cardiac transplantation.</p>","PeriodicalId":72829,"journal":{"name":"Discoveries (Craiova, Romania)","volume":"7 3","pages":"e99"},"PeriodicalIF":0.0000,"publicationDate":"2019-09-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7086075/pdf/","citationCount":"7","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Discoveries (Craiova, Romania)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.15190/d.2019.12","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 7
Abstract
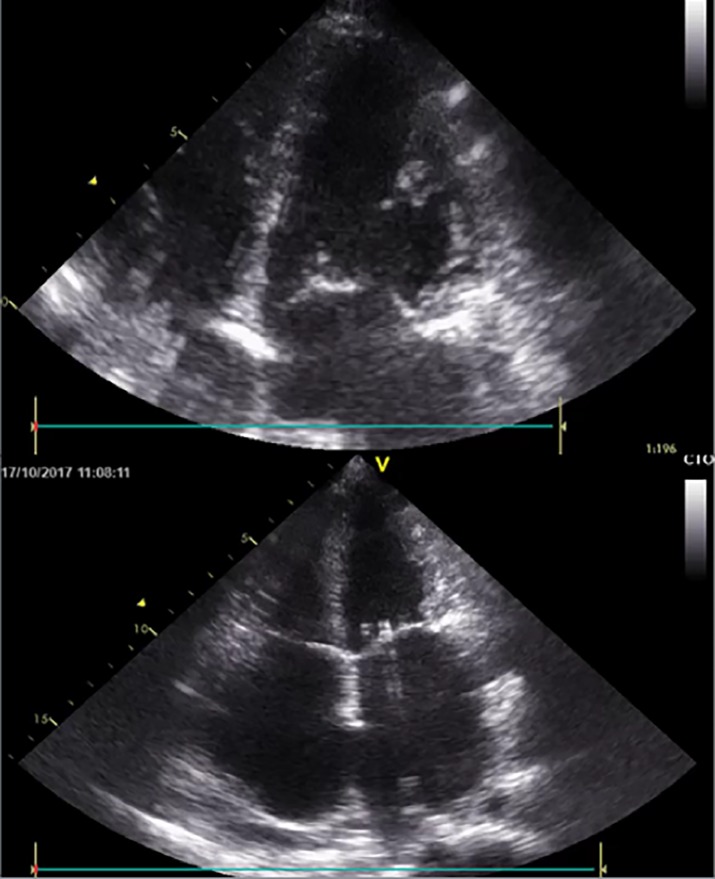
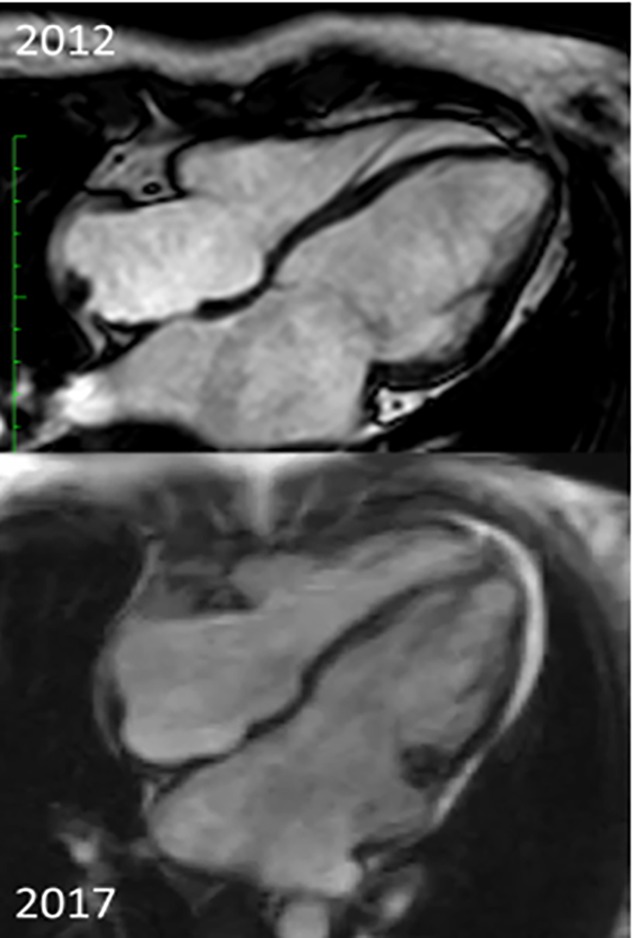
Restrictive cardiomyopathy is the least common type of cardiomyopathy, being defined by diastolic dysfunction and often unimpaired systolic function. Restrictive cardiomyopathies can be classified as familial or non-familial. Patients with familial restrictive cardiomyopathy can develop signs and symptoms of this condition anytime from childhood to adulthood. The evolution of the disease is towards signs and symptoms of pulmonary and systemic congestion and, without treatment, there is a five-year mortality rate of approximately 30% in these patients. We discuss the case of a 43-year-old patient diagnosed with familial restrictive cardiomyopathy with positive genetic tests for mutations of MYH7 gene and ABCC9 gene, who was first hospitalized in 2011 for palpitations. The echocardiography performed in evolution showed a continuous alteration of right ventricle function, without important differences of left ventricular function. She developed heart failure symptoms six years after diagnosis and she had seven hospitalizations in the past two years, currently with an increasing need of diuretics and persistent hepatic dysfunction. Cardiac transplantation or left ventricular assist device therapy should be considered in patients with severe heart failure symptoms and no longer effective treatment. However, elevated pulmonary vascular resistance excludes the patient from cardiac transplantation.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
