Shuping Qu, Qiuyuan Shi, Jing Xu, Wanwan Yi, Hengwei Fan
{"title":"Weighted Gene Coexpression Network Analysis Reveals the Dynamic Transcriptome Regulation and Prognostic Biomarkers of Hepatocellular Carcinoma.","authors":"Shuping Qu, Qiuyuan Shi, Jing Xu, Wanwan Yi, Hengwei Fan","doi":"10.1177/1176934320920562","DOIUrl":null,"url":null,"abstract":"<p><p>This study was aimed at revealing the dynamic regulation of mRNAs, long noncoding RNAs (lncRNAs), and microRNAs (miRNAs) in hepatocellular carcinoma (HCC) and to identify HCC biomarkers capable of predicting prognosis. Differentially expressed mRNAs (DEmRNAs), lncRNAs, and miRNAs were acquired by comparing expression profiles of HCC with normal samples, using an expression data set from The Cancer Genome Atlas. Altered biological functions and pathways in HCC were analyzed by subjecting DEmRNAs to Gene Ontology and Kyoto Encyclopedia of Genes and Genomes analysis. Gene modules significantly associated with disease status were identified by weighted gene coexpression network analysis. An lncRNA-mRNA and an miRNA-mRNA coexpression network were constructed for genes in disease-related modules, followed by the identification of prognostic biomarkers using Kaplan-Meier survival analysis. Differential expression and association with the prognosis of 4 miRNAs were verified in independent data sets. A total of 1220 differentially expressed genes were identified between HCC and normal samples. Differentially expressed mRNAs were significantly enriched in functions and pathways related to \"plasma membrane structure,\" \"sensory perception,\" \"metabolism,\" and \"cell proliferation.\" Two disease-associated gene modules were identified. Among genes in lncRNA-mRNA and miRNA-mRNA coexpression networks, 9 DEmRNAs and 7 DEmiRNAs were identified to be potential prognostic biomarkers. MIMAT0000102, MIMAT0003882, and MIMAT0004677 were successfully validated in independent data sets. Our results may advance our understanding of molecular mechanisms underlying HCC. The biomarkers may contribute to diagnosis in future clinical practice.</p>","PeriodicalId":50472,"journal":{"name":"Evolutionary Bioinformatics","volume":"16 ","pages":"1176934320920562"},"PeriodicalIF":1.5000,"publicationDate":"2020-05-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1177/1176934320920562","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Evolutionary Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1177/1176934320920562","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"EVOLUTIONARY BIOLOGY","Score":null,"Total":0}
引用次数: 3
Abstract
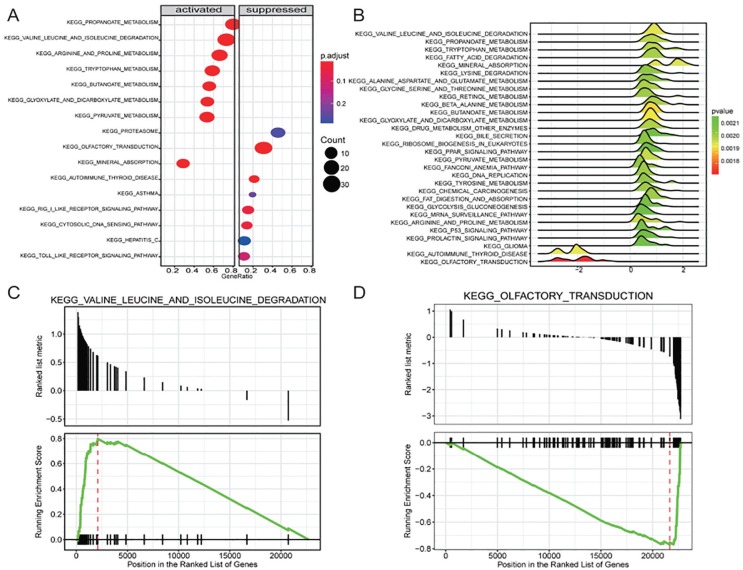
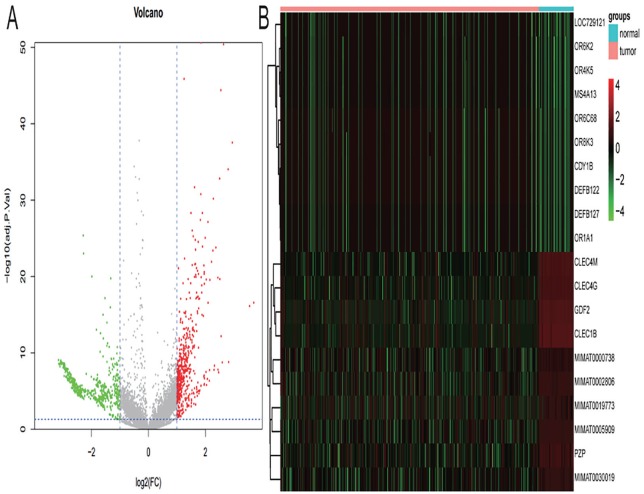
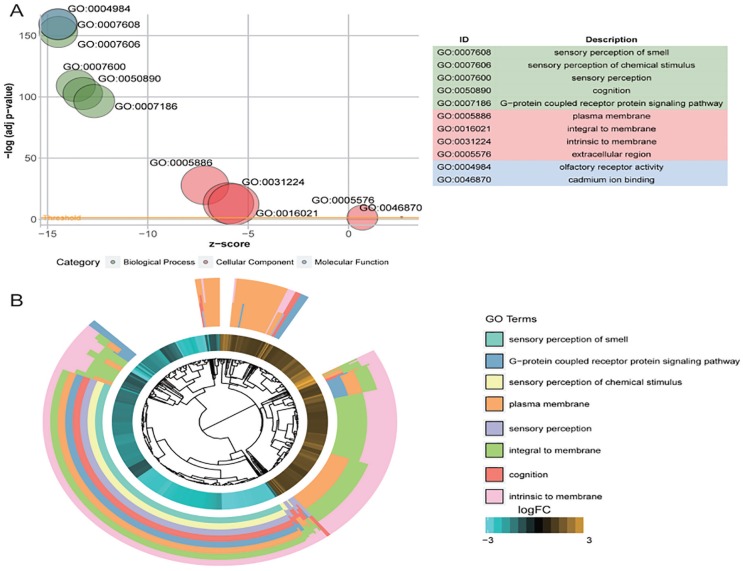
This study was aimed at revealing the dynamic regulation of mRNAs, long noncoding RNAs (lncRNAs), and microRNAs (miRNAs) in hepatocellular carcinoma (HCC) and to identify HCC biomarkers capable of predicting prognosis. Differentially expressed mRNAs (DEmRNAs), lncRNAs, and miRNAs were acquired by comparing expression profiles of HCC with normal samples, using an expression data set from The Cancer Genome Atlas. Altered biological functions and pathways in HCC were analyzed by subjecting DEmRNAs to Gene Ontology and Kyoto Encyclopedia of Genes and Genomes analysis. Gene modules significantly associated with disease status were identified by weighted gene coexpression network analysis. An lncRNA-mRNA and an miRNA-mRNA coexpression network were constructed for genes in disease-related modules, followed by the identification of prognostic biomarkers using Kaplan-Meier survival analysis. Differential expression and association with the prognosis of 4 miRNAs were verified in independent data sets. A total of 1220 differentially expressed genes were identified between HCC and normal samples. Differentially expressed mRNAs were significantly enriched in functions and pathways related to "plasma membrane structure," "sensory perception," "metabolism," and "cell proliferation." Two disease-associated gene modules were identified. Among genes in lncRNA-mRNA and miRNA-mRNA coexpression networks, 9 DEmRNAs and 7 DEmiRNAs were identified to be potential prognostic biomarkers. MIMAT0000102, MIMAT0003882, and MIMAT0004677 were successfully validated in independent data sets. Our results may advance our understanding of molecular mechanisms underlying HCC. The biomarkers may contribute to diagnosis in future clinical practice.
期刊介绍:
Evolutionary Bioinformatics is an open access, peer reviewed international journal focusing on evolutionary bioinformatics. The journal aims to support understanding of organismal form and function through use of molecular, genetic, genomic and proteomic data by giving due consideration to its evolutionary context.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
