{"title":"Mitochondria, mitophagy, and metabolic disease: towards assembling the puzzle.","authors":"Zhiyong Chen, Marine Berquez, Alessandro Luciani","doi":"10.15698/cst2020.06.222","DOIUrl":null,"url":null,"abstract":"<p><p>Dysregulation of the mitochondrial network in terminally differentiated cells contributes to a broad spectrum of disorders. Methylmalonic acidemia (MMA) is an autosomal recessive inborn error of intermediary metabolism caused by the deficiency of methylmalonyl-CoA mutase (MMUT) - a mitochondrial enzyme that mediates the degradation of certain amino acids and lipids. The loss of MMUT activity triggers an accumulation of toxic endogenous metabolites causing severe organ dysfunctions and life-threatening complications. How MMUT deficiency instigates mitochondrial distress and tissue damage remains poorly understood. Using cell and animal-based models, we recently discovered that MMUT deficiency disables the PINK1-induced translocation of PRKN/Parkin to MMA-damaged mitochondria, impeding their delivery and subsequent dismantling by macroautophagy/autophagy-lysosome degradation systems (Luciani et al. <i>Nat Commun</i>. 11(1):970). This promotes an accumulation of damaged and/or dysfunctional mitochondria that spark epithelial distress and tissue damage. Using a systems biology approach based on drug-disease network perturbation modeling, we predicted targetable pathways, whose modulation repairs mitochondrial dysfunctions in patient-derived kidney cells and ameliorates disease-relevant phenotypes in <i>mmut</i>-deficient zebrafish. These results unveil a link between primary MMUT deficiency, defective mitophagy, and cell distress, offering promising therapeutic avenues for MMA and other mitochondria-related diseases.</p>","PeriodicalId":36371,"journal":{"name":"Cell Stress","volume":"4 6","pages":"147-150"},"PeriodicalIF":3.0000,"publicationDate":"2020-05-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7278521/pdf/","citationCount":"9","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell Stress","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.15698/cst2020.06.222","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 9
Abstract
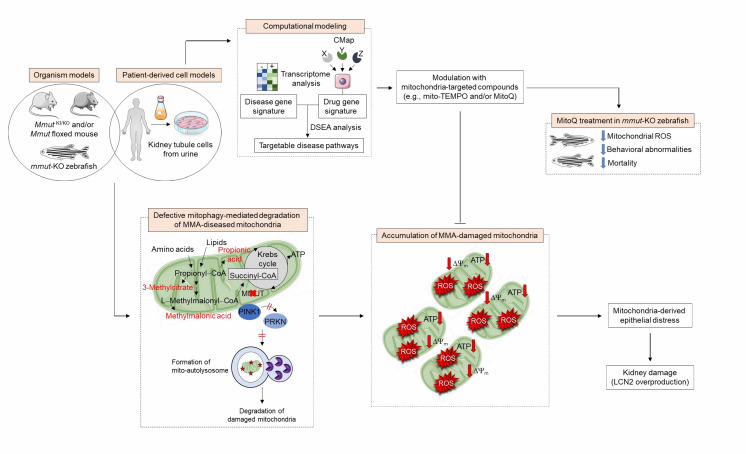
Dysregulation of the mitochondrial network in terminally differentiated cells contributes to a broad spectrum of disorders. Methylmalonic acidemia (MMA) is an autosomal recessive inborn error of intermediary metabolism caused by the deficiency of methylmalonyl-CoA mutase (MMUT) - a mitochondrial enzyme that mediates the degradation of certain amino acids and lipids. The loss of MMUT activity triggers an accumulation of toxic endogenous metabolites causing severe organ dysfunctions and life-threatening complications. How MMUT deficiency instigates mitochondrial distress and tissue damage remains poorly understood. Using cell and animal-based models, we recently discovered that MMUT deficiency disables the PINK1-induced translocation of PRKN/Parkin to MMA-damaged mitochondria, impeding their delivery and subsequent dismantling by macroautophagy/autophagy-lysosome degradation systems (Luciani et al. Nat Commun. 11(1):970). This promotes an accumulation of damaged and/or dysfunctional mitochondria that spark epithelial distress and tissue damage. Using a systems biology approach based on drug-disease network perturbation modeling, we predicted targetable pathways, whose modulation repairs mitochondrial dysfunctions in patient-derived kidney cells and ameliorates disease-relevant phenotypes in mmut-deficient zebrafish. These results unveil a link between primary MMUT deficiency, defective mitophagy, and cell distress, offering promising therapeutic avenues for MMA and other mitochondria-related diseases.
Cell StressBiochemistry, Genetics and Molecular Biology-Biochemistry, Genetics and Molecular Biology (miscellaneous)
CiteScore
13.50
自引率
0.00%
发文量
21
审稿时长
15 weeks
期刊介绍:
Cell Stress is an open-access, peer-reviewed journal that is dedicated to publishing highly relevant research in the field of cellular pathology. The journal focuses on advancing our understanding of the molecular, mechanistic, phenotypic, and other critical aspects that underpin cellular dysfunction and disease. It specifically aims to foster cell biology research that is applicable to a range of significant human diseases, including neurodegenerative disorders, myopathies, mitochondriopathies, infectious diseases, cancer, and pathological aging.
The scope of Cell Stress is broad, welcoming submissions that represent a spectrum of research from fundamental to translational and clinical studies. The journal is a valuable resource for scientists, educators, and policymakers worldwide, as well as for any individual with an interest in cellular pathology. It serves as a platform for the dissemination of research findings that are instrumental in the investigation, classification, diagnosis, and therapeutic management of major diseases. By being open-access, Cell Stress ensures that its content is freely available to a global audience, thereby promoting international scientific collaboration and accelerating the exchange of knowledge within the research community.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
