{"title":"High mitochondrial calcium levels precede neuronal death <i>in vivo</i> in Alzheimer's disease.","authors":"Maria Calvo-Rodriguez, Brian J Bacskai","doi":"10.15698/cst2020.07.226","DOIUrl":null,"url":null,"abstract":"<p><p>Alzheimer's disease (AD), the most common cause of dementia, affects millions of people worldwide. Suggested mechanisms of neurotoxicity in AD include impaired calcium (Ca<sup>2+</sup>) homeostasis and mitochondrial dysfunction, both contributing to neuronal damage. Little was known about the exact mitochondrial Ca<sup>2+</sup> homeostasis in the living brain, particularly in AD. Only now, with the development of intravital imaging techniques and transgenic mouse models of the disease, we are able to directly observe Ca<sup>2+</sup> levels in specific regions or particular subcellular compartments of cells, such as mitochondria. Using multiphoton microscopy, a Ca<sup>2+</sup> reporter targeted to mitochondria and a mouse model of cerebral β amyloidosis (APP/PS1), our recent study (Nat Comms 2020, 11:2146) found elevated mitochondrial Ca<sup>2+</sup> concentration in the transgenic mouse after plaque deposition, and after topical application of natural soluble amyloid beta (Aβ) oligomers to the healthy mouse brain at concentrations similar to those found in the human brain. Elevated Ca<sup>2+</sup> in mitochondria preceded neuronal death and could be targeted for neuroprotective therapies in AD. Here, we describe our main findings and pose new questions for future studies aimed at better understanding mitochondrial Ca<sup>2+</sup> dyshomeostasis in AD.</p>","PeriodicalId":36371,"journal":{"name":"Cell Stress","volume":"4 7","pages":"187-190"},"PeriodicalIF":3.0000,"publicationDate":"2020-06-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7328672/pdf/","citationCount":"11","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell Stress","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.15698/cst2020.07.226","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 11
Abstract
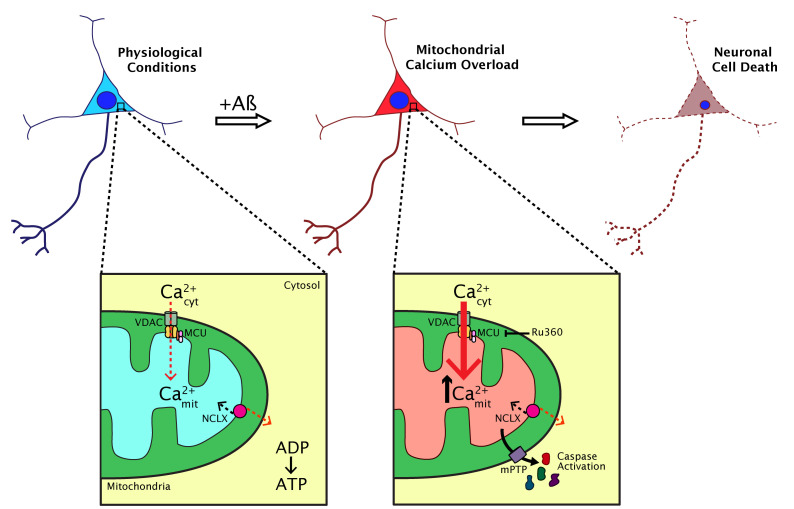
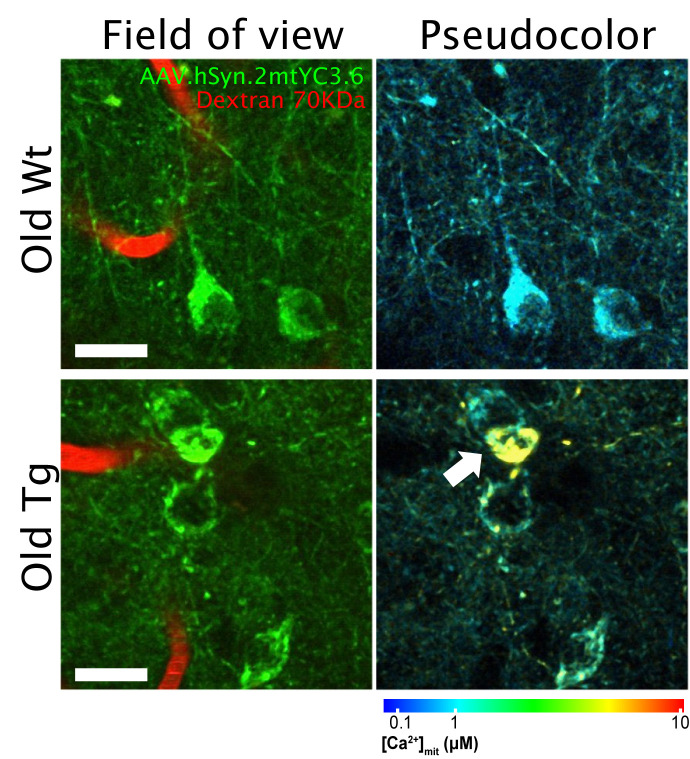
Alzheimer's disease (AD), the most common cause of dementia, affects millions of people worldwide. Suggested mechanisms of neurotoxicity in AD include impaired calcium (Ca2+) homeostasis and mitochondrial dysfunction, both contributing to neuronal damage. Little was known about the exact mitochondrial Ca2+ homeostasis in the living brain, particularly in AD. Only now, with the development of intravital imaging techniques and transgenic mouse models of the disease, we are able to directly observe Ca2+ levels in specific regions or particular subcellular compartments of cells, such as mitochondria. Using multiphoton microscopy, a Ca2+ reporter targeted to mitochondria and a mouse model of cerebral β amyloidosis (APP/PS1), our recent study (Nat Comms 2020, 11:2146) found elevated mitochondrial Ca2+ concentration in the transgenic mouse after plaque deposition, and after topical application of natural soluble amyloid beta (Aβ) oligomers to the healthy mouse brain at concentrations similar to those found in the human brain. Elevated Ca2+ in mitochondria preceded neuronal death and could be targeted for neuroprotective therapies in AD. Here, we describe our main findings and pose new questions for future studies aimed at better understanding mitochondrial Ca2+ dyshomeostasis in AD.
Cell StressBiochemistry, Genetics and Molecular Biology-Biochemistry, Genetics and Molecular Biology (miscellaneous)
CiteScore
13.50
自引率
0.00%
发文量
21
审稿时长
15 weeks
期刊介绍:
Cell Stress is an open-access, peer-reviewed journal that is dedicated to publishing highly relevant research in the field of cellular pathology. The journal focuses on advancing our understanding of the molecular, mechanistic, phenotypic, and other critical aspects that underpin cellular dysfunction and disease. It specifically aims to foster cell biology research that is applicable to a range of significant human diseases, including neurodegenerative disorders, myopathies, mitochondriopathies, infectious diseases, cancer, and pathological aging.
The scope of Cell Stress is broad, welcoming submissions that represent a spectrum of research from fundamental to translational and clinical studies. The journal is a valuable resource for scientists, educators, and policymakers worldwide, as well as for any individual with an interest in cellular pathology. It serves as a platform for the dissemination of research findings that are instrumental in the investigation, classification, diagnosis, and therapeutic management of major diseases. By being open-access, Cell Stress ensures that its content is freely available to a global audience, thereby promoting international scientific collaboration and accelerating the exchange of knowledge within the research community.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
