Shahzaib Khattak, Meryam Jan, Sara Warsi, Sohail Khattak
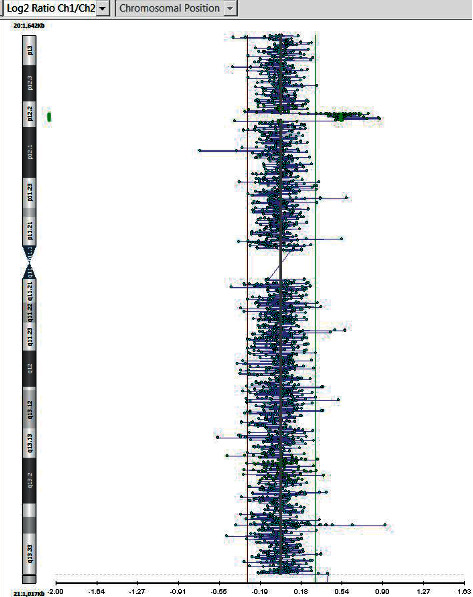
{"title":"Chromosome 20p Partial <i>De Novo</i> Duplication Identified in a Female Paediatric Patient with Characteristic Facial Dysmorphism and Behavioural Anomalies.","authors":"Shahzaib Khattak, Meryam Jan, Sara Warsi, Sohail Khattak","doi":"10.1155/2020/7093409","DOIUrl":null,"url":null,"abstract":"<p><p>Copy number variations (CNVs) involving the <i>JAG1</i> gene are rare and infrequently reported in the scientific literature. Recently, a generally healthy young patient presenting with a history of behavioural concerns was referred to us. Herein, we discuss the patient, a 7-year-old female possessing a 0.797 Mb microduplication within the short arm of chromosome 20 at band 12.2. The patient generates considerable curiosity due to the rarity of her case, which includes a <i>de novo</i> partial duplication involving the <i>JAG1</i> gene. The patient exhibits a wide range of symptoms including facial dysmorphism (dolichocephaly, round face, tented philtrum, anteverted nares, and micrognathia), clinodactyly, and an inborn congenital heart defect. She presented with behavioural concerns including ADHD-I, SPD, motor clumsiness, and poor self-regulation. Deletions in <i>JAG1</i> are often linked to <i>Alagille Syndrome</i>; however, complete duplications have not been specifically identified as disease-causing. <i>JAG1</i> mutations are reported alongside various clinical features including facial dysmorphology, heart defects, vertebral abnormalities, and ocular dysmorphic features (strabismus, epicanthal folds, and slanted palpebral fissures). This particular microduplication is rare, and thus, limited data exist regarding its significance. To our knowledge, most reported duplications are larger than 0.797 Mb. This may define a critical region causing phenotypical changes in some patient cases.</p>","PeriodicalId":30325,"journal":{"name":"Case Reports in Genetics","volume":"2020 ","pages":"7093409"},"PeriodicalIF":0.0000,"publicationDate":"2020-07-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2020/7093409","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2020/7093409","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 2
Abstract
Copy number variations (CNVs) involving the JAG1 gene are rare and infrequently reported in the scientific literature. Recently, a generally healthy young patient presenting with a history of behavioural concerns was referred to us. Herein, we discuss the patient, a 7-year-old female possessing a 0.797 Mb microduplication within the short arm of chromosome 20 at band 12.2. The patient generates considerable curiosity due to the rarity of her case, which includes a de novo partial duplication involving the JAG1 gene. The patient exhibits a wide range of symptoms including facial dysmorphism (dolichocephaly, round face, tented philtrum, anteverted nares, and micrognathia), clinodactyly, and an inborn congenital heart defect. She presented with behavioural concerns including ADHD-I, SPD, motor clumsiness, and poor self-regulation. Deletions in JAG1 are often linked to Alagille Syndrome; however, complete duplications have not been specifically identified as disease-causing. JAG1 mutations are reported alongside various clinical features including facial dysmorphology, heart defects, vertebral abnormalities, and ocular dysmorphic features (strabismus, epicanthal folds, and slanted palpebral fissures). This particular microduplication is rare, and thus, limited data exist regarding its significance. To our knowledge, most reported duplications are larger than 0.797 Mb. This may define a critical region causing phenotypical changes in some patient cases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
