D Hettiarachchi, Hetalkumar Panchal, P S Lai, V H W Dissanayake
{"title":"Novel variant in NSDHL gene associated with CHILD syndrome and syndactyly- a case report.","authors":"D Hettiarachchi, Hetalkumar Panchal, P S Lai, V H W Dissanayake","doi":"10.1186/s12881-020-01094-y","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Congenital hemidysplasia with ichthyosiform erythroderma and limb defects also known as CHILD syndrome is an X-linked dominant, male lethal genodermatosis with a prevalence of 1 in 100,000 live births. Mutations in NSDHL gene located at Xq28 potentially impair the function of NAD(P) H steroid dehydrogenase-like protein and is responsible for its pathogenesis.</p><p><strong>Case presentation: </strong>The proband was a 9-month-old twin (T2) girl with a healthy twin sister (T1) of Sri Lankan origin born to non-consanguineous parents. She presented with right sided continuous icthyosiform erythroderma and ipsilateral limb defects and congenital hemidysplasia since birth. Notably the child had ipsilateral hand hypoplasia and syndactyly. There were other visceral abnormalities. We performed whole exome sequencing and found a novel heterozygous variant (NSDHL, c.713C > A, p.Thr238Asn).</p><p><strong>Conclusion: </strong>We report a novel missense variant in the NSDHL gene that resides in a highly-conserved region. This variant affects the NAD(P) H steroid dehydrogenase-like protein function via reduction in the number of active sites resulting in the CHILD syndrome phenotype and syndactyly.</p>","PeriodicalId":9015,"journal":{"name":"BMC Medical Genetics","volume":" ","pages":"164"},"PeriodicalIF":0.0000,"publicationDate":"2020-08-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12881-020-01094-y","citationCount":"7","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12881-020-01094-y","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 7
Abstract
Background: Congenital hemidysplasia with ichthyosiform erythroderma and limb defects also known as CHILD syndrome is an X-linked dominant, male lethal genodermatosis with a prevalence of 1 in 100,000 live births. Mutations in NSDHL gene located at Xq28 potentially impair the function of NAD(P) H steroid dehydrogenase-like protein and is responsible for its pathogenesis.
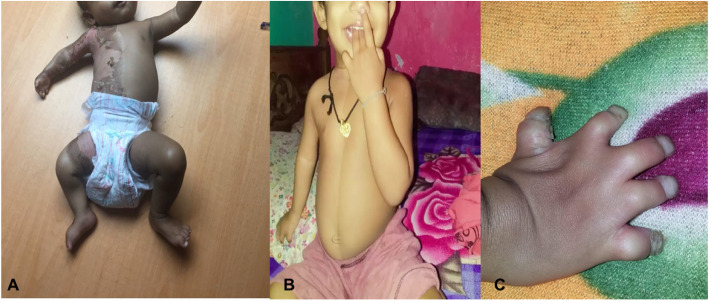
Case presentation: The proband was a 9-month-old twin (T2) girl with a healthy twin sister (T1) of Sri Lankan origin born to non-consanguineous parents. She presented with right sided continuous icthyosiform erythroderma and ipsilateral limb defects and congenital hemidysplasia since birth. Notably the child had ipsilateral hand hypoplasia and syndactyly. There were other visceral abnormalities. We performed whole exome sequencing and found a novel heterozygous variant (NSDHL, c.713C > A, p.Thr238Asn).
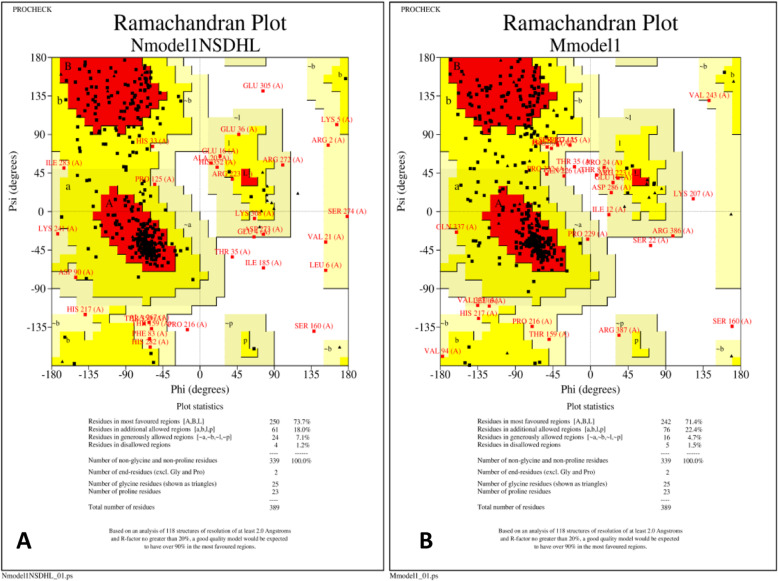
Conclusion: We report a novel missense variant in the NSDHL gene that resides in a highly-conserved region. This variant affects the NAD(P) H steroid dehydrogenase-like protein function via reduction in the number of active sites resulting in the CHILD syndrome phenotype and syndactyly.
期刊介绍:
BMC Medical Genetics is an open access journal publishing original peer-reviewed research articles in the effects of genetic variation in individuals, families and among populations in relation to human health and disease.
Note: BMC Medical Genetics is now closed. This journal has merged with BMC Medical Genomics, a broad-scope, open access community journal for all medical genetics and genomics research.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
