Francesca Magri, Roberta Brusa, Luca Bello, Lorenzo Peverelli, Roberto Del Bo, Alessandra Govoni, Claudia Cinnante, Irene Colombo, Francesco Fortunato, Roberto Tironi, Stefania Corti, Nadia Grimoldi, Monica Sciacco, Nereo Bresolin, Elena Pegoraro, Maurizio Moggio, Giacomo Pietro Comi
{"title":"Limb girdle muscular dystrophy due to <i>LAMA2</i> gene mutations: new mutations expand the clinical spectrum of a still challenging diagnosis.","authors":"Francesca Magri, Roberta Brusa, Luca Bello, Lorenzo Peverelli, Roberto Del Bo, Alessandra Govoni, Claudia Cinnante, Irene Colombo, Francesco Fortunato, Roberto Tironi, Stefania Corti, Nadia Grimoldi, Monica Sciacco, Nereo Bresolin, Elena Pegoraro, Maurizio Moggio, Giacomo Pietro Comi","doi":"10.36185/2532-1900-009","DOIUrl":null,"url":null,"abstract":"<p><p>Mutations in <i>LAMA2</i> gene, encoding merosin, are generally responsible of a severe congenital-onset muscular dystrophy (CMD type 1A) characterized by severe weakness, merosin absence at muscle analysis and white matter alterations at brain Magnetic Resonance Imaging (MRI). Recently, <i>LAMA2</i> mutations have been acknowledged as responsible of LGMD R23, despite only few cases with slowly progressive adult-onset and partial merosin deficiency have been reported. We describe 5 independent Italian subjects presenting with progressive limb girdle muscular weakness, brain white matter abnormalities, merosin deficiency and <i>LAMA2</i> gene mutations. We detected 7 different mutations, 6 of which are new. All patients showed normal psicomotor development and slowly progressive weakness with onset spanning from childhood to forties. Creatin-kinase levels were moderately elevated. One patient showed dilated cardiomyopathy. Muscle MRI allowed to evaluate the degree and pattern of muscular involvement in all patients. Brain MRI was fundamental in order to address and/or support the molecular diagnosis, showing typical widespread white matter hyperintensity in T2-weighted sequences. Interestingly these alterations were associated with central nervous system involvement in 3 patients who presented epilepsy and migraine. Muscle biopsy commonly but not necessarily revealed dystrophic features. Western-blot was usually more accurate than immunohystochemical analysis in detecting merosin deficiency. The description of these cases further enlarges the clinical spectrum of <i>LAMA2</i>-related disorders. Moreover, it supports the inclusion of LGMD R23 in the new classification of LGMD. The central nervous system involvement was fundamental to address the diagnosis and should be always included in the diagnostic work-up of undiagnosed LGMD.</p>","PeriodicalId":35953,"journal":{"name":"Acta Myologica","volume":"39 2","pages":"67-82"},"PeriodicalIF":0.0000,"publicationDate":"2020-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/0f/b8/am-2020-02-67.PMC7460730.pdf","citationCount":"5","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Myologica","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.36185/2532-1900-009","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 5
Abstract
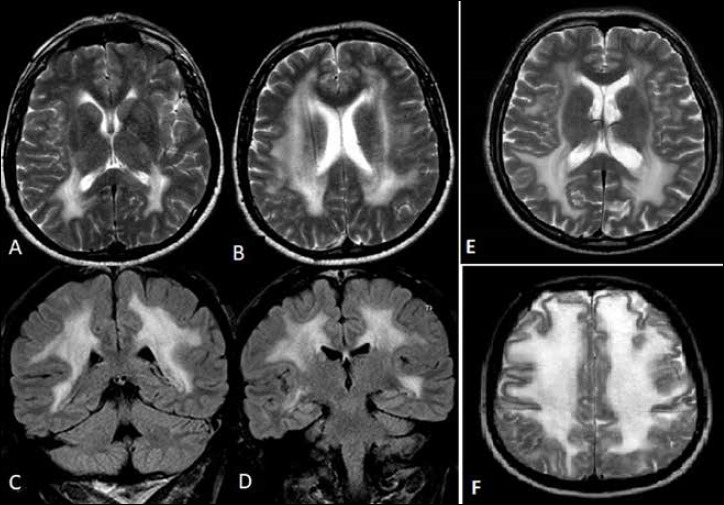
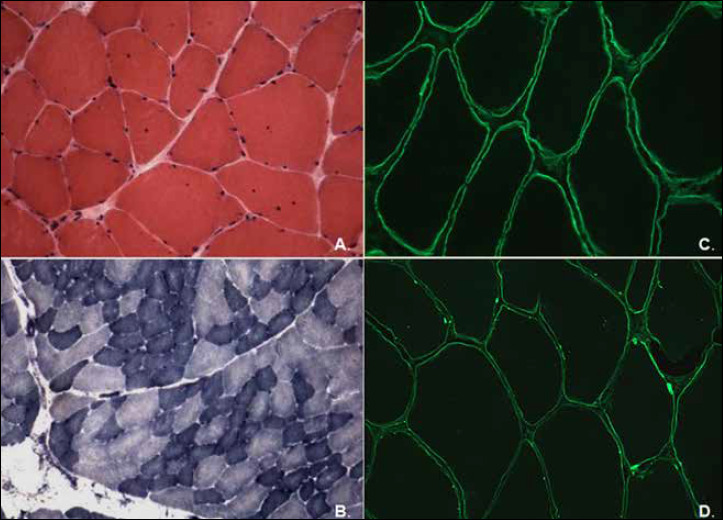
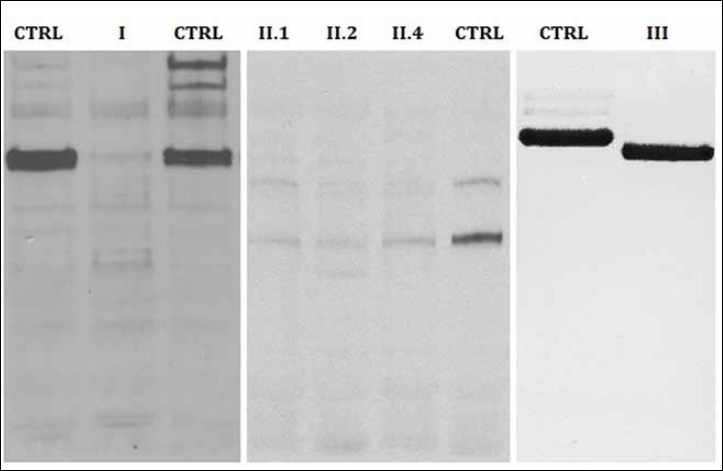
Mutations in LAMA2 gene, encoding merosin, are generally responsible of a severe congenital-onset muscular dystrophy (CMD type 1A) characterized by severe weakness, merosin absence at muscle analysis and white matter alterations at brain Magnetic Resonance Imaging (MRI). Recently, LAMA2 mutations have been acknowledged as responsible of LGMD R23, despite only few cases with slowly progressive adult-onset and partial merosin deficiency have been reported. We describe 5 independent Italian subjects presenting with progressive limb girdle muscular weakness, brain white matter abnormalities, merosin deficiency and LAMA2 gene mutations. We detected 7 different mutations, 6 of which are new. All patients showed normal psicomotor development and slowly progressive weakness with onset spanning from childhood to forties. Creatin-kinase levels were moderately elevated. One patient showed dilated cardiomyopathy. Muscle MRI allowed to evaluate the degree and pattern of muscular involvement in all patients. Brain MRI was fundamental in order to address and/or support the molecular diagnosis, showing typical widespread white matter hyperintensity in T2-weighted sequences. Interestingly these alterations were associated with central nervous system involvement in 3 patients who presented epilepsy and migraine. Muscle biopsy commonly but not necessarily revealed dystrophic features. Western-blot was usually more accurate than immunohystochemical analysis in detecting merosin deficiency. The description of these cases further enlarges the clinical spectrum of LAMA2-related disorders. Moreover, it supports the inclusion of LGMD R23 in the new classification of LGMD. The central nervous system involvement was fundamental to address the diagnosis and should be always included in the diagnostic work-up of undiagnosed LGMD.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
