{"title":"Proteinuria as a presenting sign of combined methylmalonic acidemia and homocysteinemia: case report.","authors":"Ru-Yue Chen, Xiao-Zhong Li, Qiang Lin, Yun Zhu, Yun-Yan Shen, Qin-Ying Xu, Xue-Ming Zhu, Lin-Qi Chen, Hai-Ying Wu, Xu-Qin Chen","doi":"10.1186/s12881-020-01122-x","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Disorders of the metabolism and absorption of vitamin B12 can lead to decrease in activity of methionine synthetase and methylmalonate coenzyme A mutase (MMUT), which results in increased levels of methylmalonic acid and homocysteine in blood and urine. Often, combined methylmalonic acidemia (MMA) and homocysteinemia is misdiagnosed due to a lack of specific symptoms. The clinical manifestations are diverse, but proteinuria as the initial presentation is rare.</p><p><strong>Case presentation: </strong>Two cases of MMA with homocysteinemia in children are reported. Proteinuria were a primary presenting symptom, followed by anemia and neurologic symptoms (frequent convulsions and unstable walking, respectively). Screening of amino acids and acyl carnitine in serum showed that the propionyl carnitine:acetylcarnitine ratio increased. Profiling of urinary organic acids by gas chromatography-mass spectrometry revealed high levels of methylmalonic acid. Homocysteine content in blood was increased. Comprehensive genetic analyses of peripheral blood-derived DNA demonstrated heterozygous variants of methylmalonic aciduria type C and homocystinuria (MMACHC) and amnionless (AMN) genes in our two patients, respectively. After active treatment, the clinical manifestations in Case 1 were relieved and urinary protein ceased to be observed; Case 2 had persistent proteinuria and was lost to follow-up.</p><p><strong>Conclusions: </strong>Analyses of the organic acids in blood and urine suggested MMA combined with homocysteinemia. In such diseases, reports of renal damage are uncommon and proteinuria as the initial presentation is rare. Molecular analysis indicated two different genetic causes. Although the pathologic mechanisms were related to vitamin B12, the severity and prognosis of renal lesions were different. Therefore, gene detection provides new insights into inherited metabolic diseases.</p>","PeriodicalId":9015,"journal":{"name":"BMC Medical Genetics","volume":" ","pages":"183"},"PeriodicalIF":0.0000,"publicationDate":"2020-09-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12881-020-01122-x","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12881-020-01122-x","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 4
Abstract
Background: Disorders of the metabolism and absorption of vitamin B12 can lead to decrease in activity of methionine synthetase and methylmalonate coenzyme A mutase (MMUT), which results in increased levels of methylmalonic acid and homocysteine in blood and urine. Often, combined methylmalonic acidemia (MMA) and homocysteinemia is misdiagnosed due to a lack of specific symptoms. The clinical manifestations are diverse, but proteinuria as the initial presentation is rare.
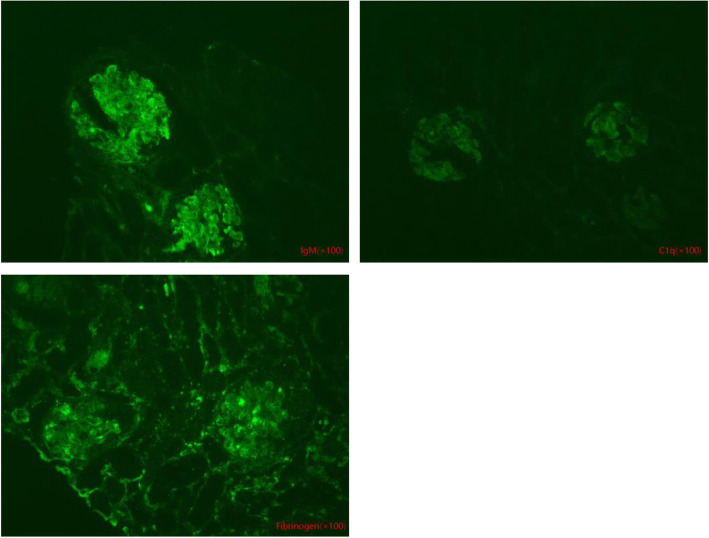
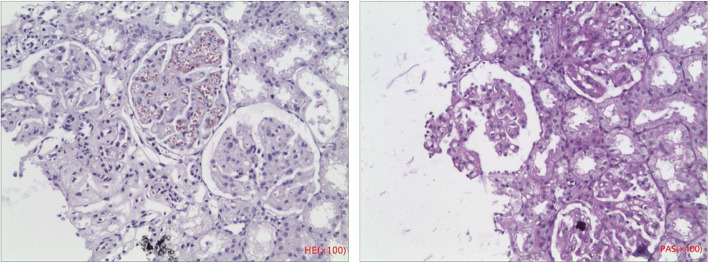
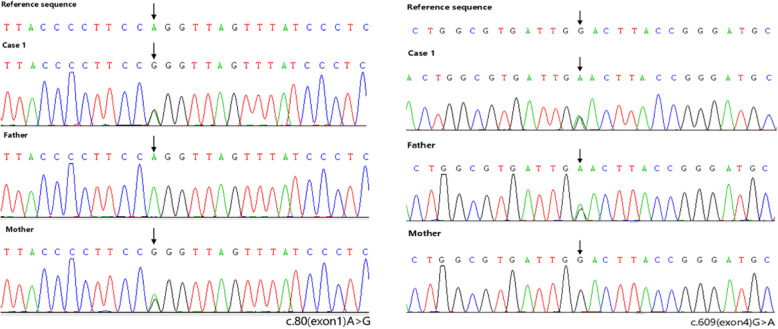
Case presentation: Two cases of MMA with homocysteinemia in children are reported. Proteinuria were a primary presenting symptom, followed by anemia and neurologic symptoms (frequent convulsions and unstable walking, respectively). Screening of amino acids and acyl carnitine in serum showed that the propionyl carnitine:acetylcarnitine ratio increased. Profiling of urinary organic acids by gas chromatography-mass spectrometry revealed high levels of methylmalonic acid. Homocysteine content in blood was increased. Comprehensive genetic analyses of peripheral blood-derived DNA demonstrated heterozygous variants of methylmalonic aciduria type C and homocystinuria (MMACHC) and amnionless (AMN) genes in our two patients, respectively. After active treatment, the clinical manifestations in Case 1 were relieved and urinary protein ceased to be observed; Case 2 had persistent proteinuria and was lost to follow-up.
Conclusions: Analyses of the organic acids in blood and urine suggested MMA combined with homocysteinemia. In such diseases, reports of renal damage are uncommon and proteinuria as the initial presentation is rare. Molecular analysis indicated two different genetic causes. Although the pathologic mechanisms were related to vitamin B12, the severity and prognosis of renal lesions were different. Therefore, gene detection provides new insights into inherited metabolic diseases.
期刊介绍:
BMC Medical Genetics is an open access journal publishing original peer-reviewed research articles in the effects of genetic variation in individuals, families and among populations in relation to human health and disease.
Note: BMC Medical Genetics is now closed. This journal has merged with BMC Medical Genomics, a broad-scope, open access community journal for all medical genetics and genomics research.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
