Peter Sparber, Margarita Sharova, Alexandra Filatova, Olga Shchagina, Evgeniya Ivanova, Elena Dadali, Mikhail Skoblov
{"title":"Recessive myotonia congenita caused by a homozygous splice site variant in CLCN1 gene: a case report.","authors":"Peter Sparber, Margarita Sharova, Alexandra Filatova, Olga Shchagina, Evgeniya Ivanova, Elena Dadali, Mikhail Skoblov","doi":"10.1186/s12881-020-01128-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Myotonia congenita is a rare neuromuscular disease, which is characterized by a delay in muscle relaxation after evoked or voluntary contraction. Myotonia congenita can be inherited in a dominant (Thomsen disease) and recessive form (Becker disease) and both are caused by pathogenic variants in the CLCN1 gene. Noncanonical splice site variants are often classified as variants of uncertain significance, due to insufficient accuracy of splice-predicting tools. Functional analysis using minigene plasmids is widely used in such cases. Moreover, functional analysis is very useful in investigation of the disease pathogenesis, which is necessary for development of future therapeutic approaches. To our knowledge only one noncanonical splice site variant in the CLCN1 gene was functionally characterized to date. We further contribute to this field by evaluation the molecular mechanism of splicing alteration caused by the c.1582 + 5G > A in a homozygous state.</p><p><strong>Case presentation: </strong>We report a clinical case of an affected 6-y.o boy with athletic appearance due to muscle hypertrophy, calf muscle stiffness, cramping and various myotonic signs in a consanguineous family with no history of neuromuscular disorders. The neurological examination showed percussion-activated myotonia in the hands and legs. Plasma creatine kinase enzyme and transaminases levels were normal. Electromyography at the time of examination shows myotonic runs in the upper and lower extremities.</p><p><strong>Conclusions: </strong>Functional analysis of the variant in a minigene system showed alteration of splicing leading to loss of function, thereby confirming that the variant is pathogenic.</p>","PeriodicalId":9015,"journal":{"name":"BMC Medical Genetics","volume":"21 Suppl 1","pages":"197"},"PeriodicalIF":0.0000,"publicationDate":"2020-10-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12881-020-01128-5","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12881-020-01128-5","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 4
Abstract
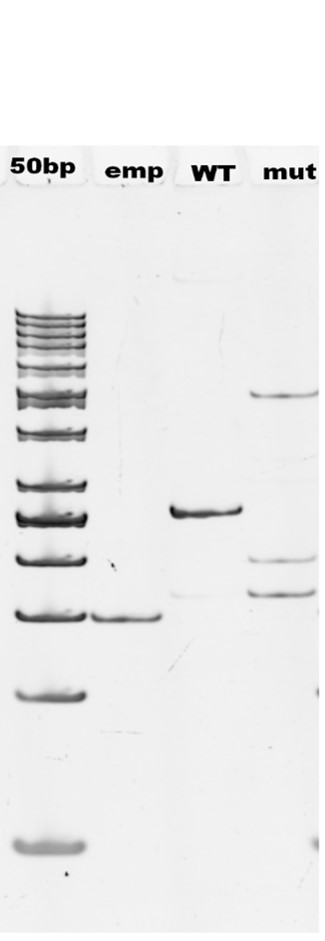
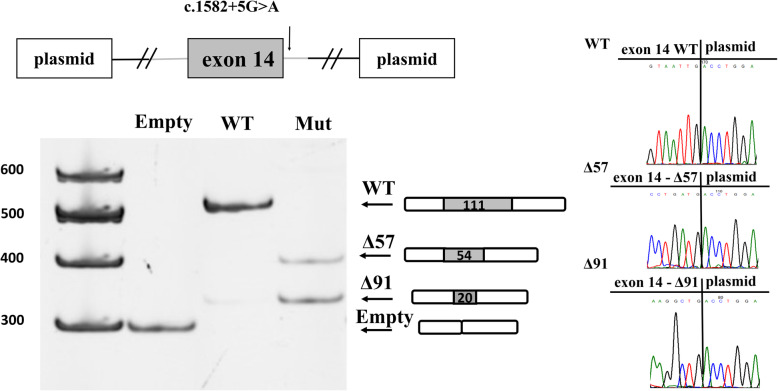
Background: Myotonia congenita is a rare neuromuscular disease, which is characterized by a delay in muscle relaxation after evoked or voluntary contraction. Myotonia congenita can be inherited in a dominant (Thomsen disease) and recessive form (Becker disease) and both are caused by pathogenic variants in the CLCN1 gene. Noncanonical splice site variants are often classified as variants of uncertain significance, due to insufficient accuracy of splice-predicting tools. Functional analysis using minigene plasmids is widely used in such cases. Moreover, functional analysis is very useful in investigation of the disease pathogenesis, which is necessary for development of future therapeutic approaches. To our knowledge only one noncanonical splice site variant in the CLCN1 gene was functionally characterized to date. We further contribute to this field by evaluation the molecular mechanism of splicing alteration caused by the c.1582 + 5G > A in a homozygous state.
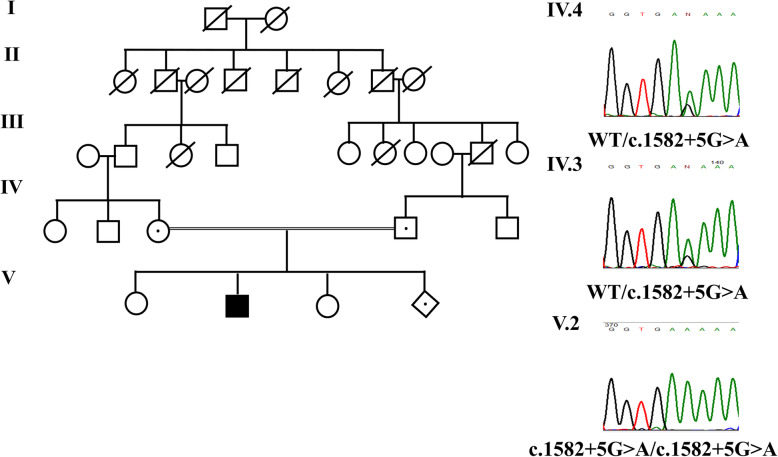
Case presentation: We report a clinical case of an affected 6-y.o boy with athletic appearance due to muscle hypertrophy, calf muscle stiffness, cramping and various myotonic signs in a consanguineous family with no history of neuromuscular disorders. The neurological examination showed percussion-activated myotonia in the hands and legs. Plasma creatine kinase enzyme and transaminases levels were normal. Electromyography at the time of examination shows myotonic runs in the upper and lower extremities.
Conclusions: Functional analysis of the variant in a minigene system showed alteration of splicing leading to loss of function, thereby confirming that the variant is pathogenic.
期刊介绍:
BMC Medical Genetics is an open access journal publishing original peer-reviewed research articles in the effects of genetic variation in individuals, families and among populations in relation to human health and disease.
Note: BMC Medical Genetics is now closed. This journal has merged with BMC Medical Genomics, a broad-scope, open access community journal for all medical genetics and genomics research.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
