G N Cerbino, L Abou Assali, L S Varela, L Tomassi, A Batlle, V E Parera, M V Rossetti
{"title":"Acute Intermittent Porphyria in a Man with Dual Enzyme Deficiencies.","authors":"G N Cerbino, L Abou Assali, L S Varela, L Tomassi, A Batlle, V E Parera, M V Rossetti","doi":"10.1155/2020/8873219","DOIUrl":null,"url":null,"abstract":"<p><p>Porphyrias are a heterogeneous group of metabolic disorders that result from the altered activity of specific enzymes of the heme biosynthetic pathway and are characterized by accumulation of pathway intermediates. Porphyria cutanea tarda (PCT) is the most common porphyria and is due to deficient activity of uroporphyrinogen decarboxylase (UROD). Acute intermittent porphyria (AIP) is the most common of the acute hepatic porphyrias, caused by decreased activity of hydroxymethylbilane synthase (HMBS). An Argentinean man with a family history of PCT who carried the <i>UROD</i> variant c.10_11insA suffered severe abdominal pain. Biochemical testing was consistent with AIP, and molecular analysis of <i>HMBS</i> revealed a <i>de novo</i> variant: c.344 + 2_ + 5delTAAG. This is one of the few cases of porphyria identified with both <i>UROD</i> and <i>HMBS</i> mutations and the first confirmed case of porphyria with dual enzyme deficiencies in Argentina.</p>","PeriodicalId":30325,"journal":{"name":"Case Reports in Genetics","volume":"2020 ","pages":"8873219"},"PeriodicalIF":0.0000,"publicationDate":"2020-10-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2020/8873219","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2020/8873219","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 1
Abstract
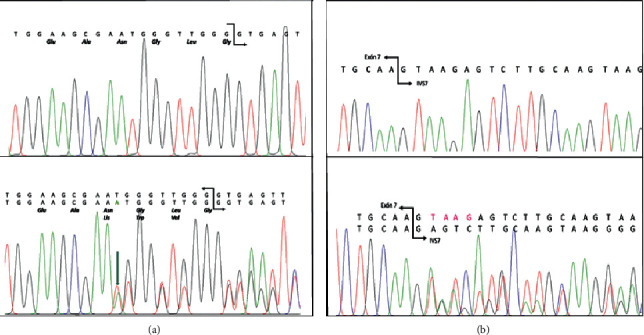
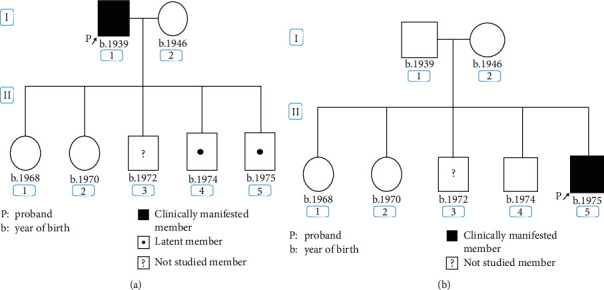
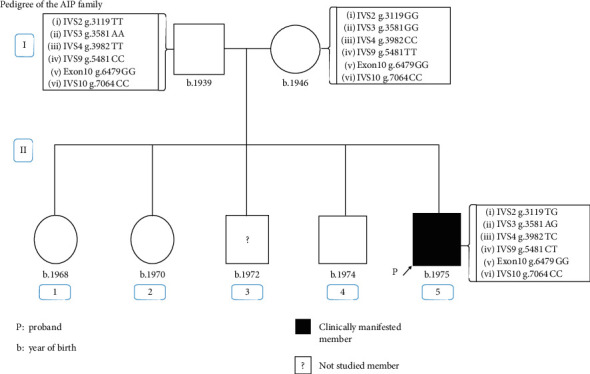
Porphyrias are a heterogeneous group of metabolic disorders that result from the altered activity of specific enzymes of the heme biosynthetic pathway and are characterized by accumulation of pathway intermediates. Porphyria cutanea tarda (PCT) is the most common porphyria and is due to deficient activity of uroporphyrinogen decarboxylase (UROD). Acute intermittent porphyria (AIP) is the most common of the acute hepatic porphyrias, caused by decreased activity of hydroxymethylbilane synthase (HMBS). An Argentinean man with a family history of PCT who carried the UROD variant c.10_11insA suffered severe abdominal pain. Biochemical testing was consistent with AIP, and molecular analysis of HMBS revealed a de novo variant: c.344 + 2_ + 5delTAAG. This is one of the few cases of porphyria identified with both UROD and HMBS mutations and the first confirmed case of porphyria with dual enzyme deficiencies in Argentina.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
