Senmao Chai, Rong Jiao, Xiaodong Sun, Pan Fu, Qiang Zhao, Ming Sang
{"title":"Novel nonsense mutation p. Gln264Ter in the ANK1 confirms causative role for hereditary spherocytosis: a case report.","authors":"Senmao Chai, Rong Jiao, Xiaodong Sun, Pan Fu, Qiang Zhao, Ming Sang","doi":"10.1186/s12881-020-01161-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Hereditary spherocytosis (HS) is the most common haemolytic anaemia caused by congenital membrane defects of red blood cells. The name derives from the presence of spherical red blood cells in the peripheral blood. Clinical manifestations of HS are anaemia, haemolytic jaundice, and large spleen, and infection can worsen the condition, often with cholelithiasis. HS is mainly caused by abnormal functions of the products of six genes. Splenectomy is the main treatment for HS.</p><p><strong>Case presentation: </strong>Half a day after birth, the proband exhibited HS-related symptoms, with progressive aggravation. Routine examination in the outpatient department showed an increase in white blood cells and a decrease in red blood cells. His mother had HS and a partial splenectomy. We suspected that the infant might also have HS. Genomic DNA samples were extracted from the three members of the HS trio pedigree, and genomic whole-exome sequencing (WES) was performed. The three DNA samples were amplified by polymerase chain reaction (PCR), followed by Sanger sequencing to identify mutation sites. A novel nonsense heterozygous mutation, c.790C > T (p. Gln264Ter), in the ANK1 gene, which causes premature termination of translation, was found in this Chinese family with autosomal dominant HS.</p><p><strong>Conclusions: </strong>This de novo nonsense mutation can cause the onset of HS in early childhood, with severe symptoms. Expanding the ANK1 genotype mutation spectrum will lay a foundation for the further application of mutation screening in genetic counselling.</p>","PeriodicalId":9015,"journal":{"name":"BMC Medical Genetics","volume":" ","pages":"223"},"PeriodicalIF":0.0000,"publicationDate":"2020-11-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12881-020-01161-4","citationCount":"5","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12881-020-01161-4","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 5
Abstract
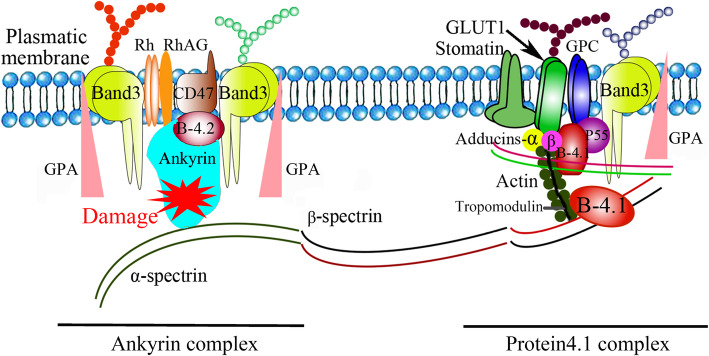
Background: Hereditary spherocytosis (HS) is the most common haemolytic anaemia caused by congenital membrane defects of red blood cells. The name derives from the presence of spherical red blood cells in the peripheral blood. Clinical manifestations of HS are anaemia, haemolytic jaundice, and large spleen, and infection can worsen the condition, often with cholelithiasis. HS is mainly caused by abnormal functions of the products of six genes. Splenectomy is the main treatment for HS.
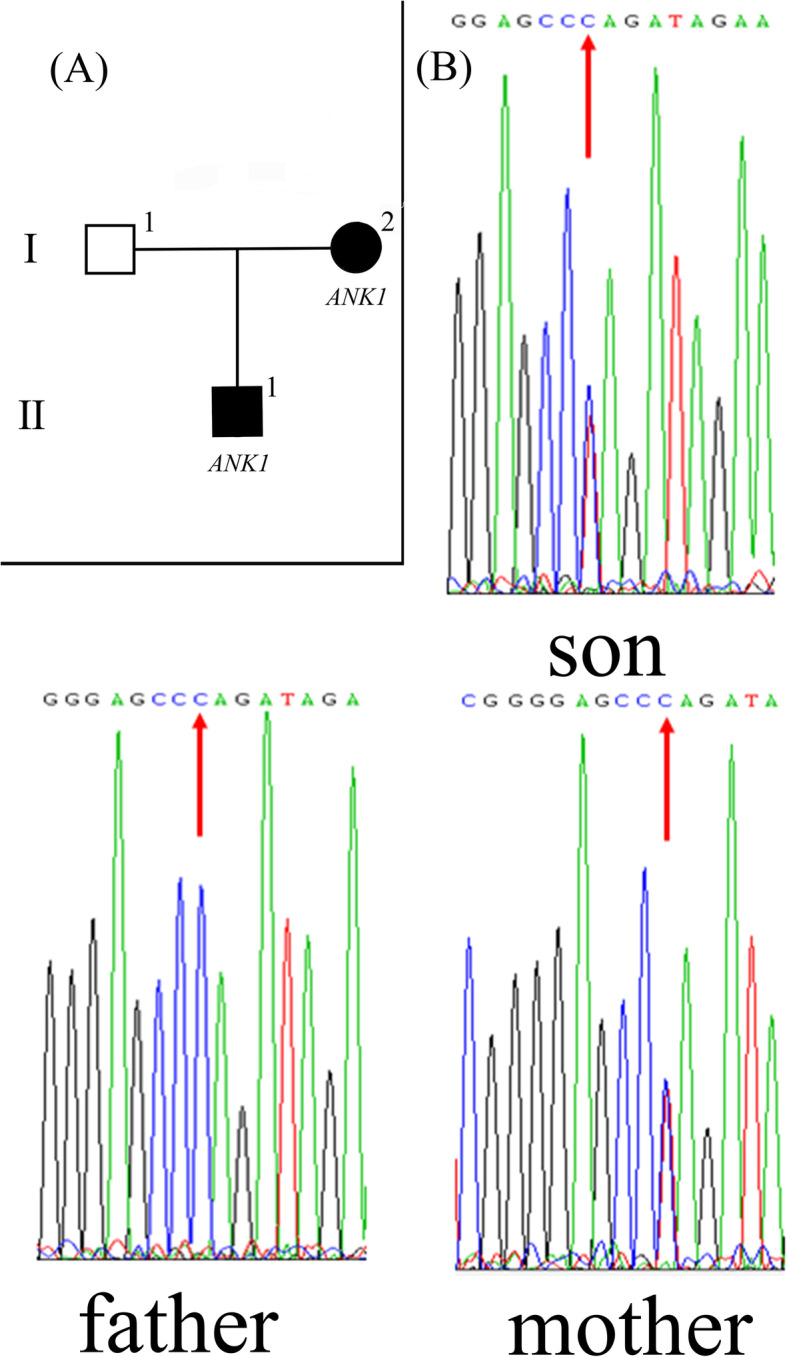
Case presentation: Half a day after birth, the proband exhibited HS-related symptoms, with progressive aggravation. Routine examination in the outpatient department showed an increase in white blood cells and a decrease in red blood cells. His mother had HS and a partial splenectomy. We suspected that the infant might also have HS. Genomic DNA samples were extracted from the three members of the HS trio pedigree, and genomic whole-exome sequencing (WES) was performed. The three DNA samples were amplified by polymerase chain reaction (PCR), followed by Sanger sequencing to identify mutation sites. A novel nonsense heterozygous mutation, c.790C > T (p. Gln264Ter), in the ANK1 gene, which causes premature termination of translation, was found in this Chinese family with autosomal dominant HS.
Conclusions: This de novo nonsense mutation can cause the onset of HS in early childhood, with severe symptoms. Expanding the ANK1 genotype mutation spectrum will lay a foundation for the further application of mutation screening in genetic counselling.
背景:遗传性球形红细胞增多症(HS)是由先天性红细胞膜缺陷引起的最常见的溶血性贫血。这个名字来源于外周血中球形红细胞的存在。HS的临床表现为贫血、溶血性黄疸和脾大,感染可使病情恶化,常伴有胆石症。HS主要由6种基因产物的功能异常引起。脾切除术是治疗HS的主要方法。病例介绍:先证者出生后半天出现hs相关症状,并逐渐加重。门诊常规检查显示白细胞增多,红细胞减少。他母亲患了HS,还切除了部分脾。我们怀疑婴儿也可能患有HS。从HS三人家系的三个成员中提取基因组DNA样本,并进行基因组全外显子组测序(WES)。采用聚合酶链反应(PCR)扩增3份DNA样本,然后进行Sanger测序以确定突变位点。在这个常染色体显性HS的中国家庭中发现了ANK1基因中一种新的无义杂合突变,c.790C > T (p. Gln264Ter),可导致翻译过早终止。结论:这种从头无义突变可导致儿童期早期HS发病,症状严重。扩大ANK1基因型突变谱,为突变筛查在遗传咨询中的进一步应用奠定基础。
期刊介绍:
BMC Medical Genetics is an open access journal publishing original peer-reviewed research articles in the effects of genetic variation in individuals, families and among populations in relation to human health and disease.
Note: BMC Medical Genetics is now closed. This journal has merged with BMC Medical Genomics, a broad-scope, open access community journal for all medical genetics and genomics research.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
