Clemens V Farr, Ali El-Kasaby, Michael Freissmuth, Sonja Sucic
{"title":"The Creatine Transporter Unfolded: A Knotty Premise in the Cerebral Creatine Deficiency Syndrome.","authors":"Clemens V Farr, Ali El-Kasaby, Michael Freissmuth, Sonja Sucic","doi":"10.3389/fnsyn.2020.588954","DOIUrl":null,"url":null,"abstract":"<p><p>Creatine provides cells with high-energy phosphates for the rapid reconstitution of hydrolyzed adenosine triphosphate. The eponymous creatine transporter (CRT1/SLC6A8) belongs to a family of solute carrier 6 (SLC6) proteins. The key role of CRT1 is to translocate creatine across tissue barriers and into target cells, such as neurons and myocytes. Individuals harboring mutations in the coding sequence of the human CRT1 gene develop creatine transporter deficiency (CTD), one of the pivotal underlying causes of cerebral creatine deficiency syndrome. CTD encompasses an array of clinical manifestations, including severe intellectual disability, epilepsy, autism, development delay, and motor dysfunction. CTD is characterized by the absence of cerebral creatine, which implies an indispensable role for CRT1 in supplying the brain cells with creatine. CTD-associated variants dramatically reduce or abolish creatine transport activity by CRT1. Many of these are point mutations that are known to trigger folding defects, leading to the retention of encoded CRT1 proteins in the endoplasmic reticulum and precluding their delivery to the cell surface. Misfolding of several related SLC6 transporters also gives rise to detrimental pathologic conditions in people; e.g., mutations in the dopamine transporter induce infantile parkinsonism/dystonia, while mutations in the GABA transporter 1 cause treatment-resistant epilepsy. In some cases, folding defects are amenable to rescue by small molecules, known as pharmacological and chemical chaperones, which restore the cell surface expression and transport activity of the previously non-functional proteins. Insights from the recent molecular, animal and human case studies of CTD add toward our understanding of this complex disorder and reveal the wide-ranging effects elicited upon CRT1 dysfunction. This grants novel therapeutic prospects for the treatment of patients afflicted with CTD, e.g., modifying the creatine molecule to facilitate CRT1-independent entry into brain cells, or correcting folding-deficient and loss-of-function CTD variants using pharmacochaperones and/or allosteric modulators. The latter justifies a search for additional compounds with a capacity to correct mutation-specific defects.</p>","PeriodicalId":12650,"journal":{"name":"Frontiers in Synaptic Neuroscience","volume":"12 ","pages":"588954"},"PeriodicalIF":4.1000,"publicationDate":"2020-10-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7644880/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in Synaptic Neuroscience","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.3389/fnsyn.2020.588954","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
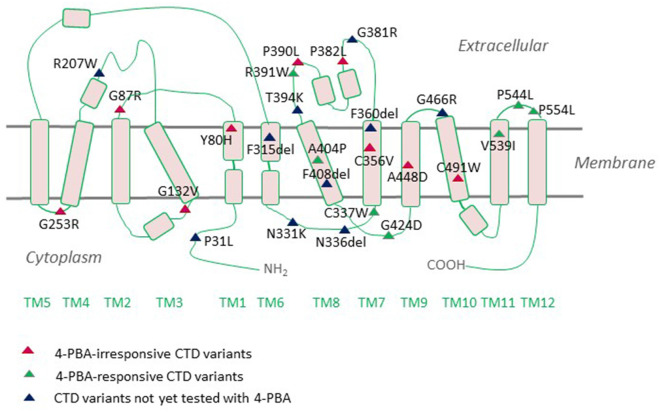
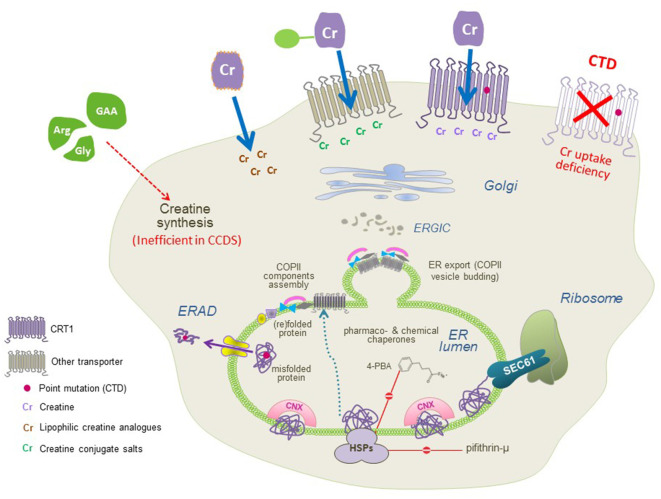
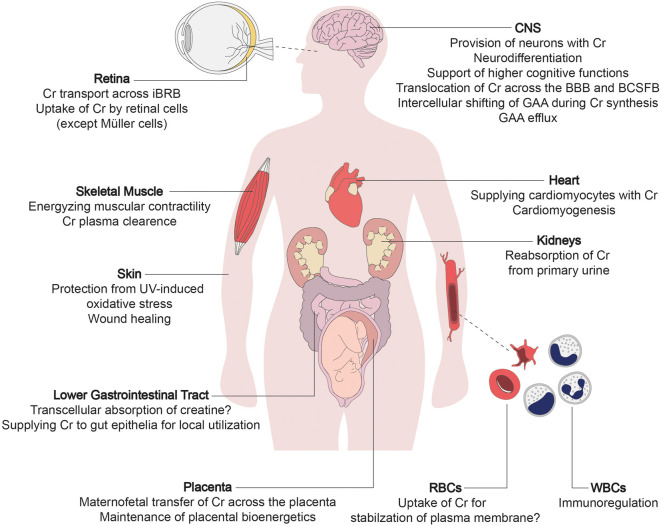
Creatine provides cells with high-energy phosphates for the rapid reconstitution of hydrolyzed adenosine triphosphate. The eponymous creatine transporter (CRT1/SLC6A8) belongs to a family of solute carrier 6 (SLC6) proteins. The key role of CRT1 is to translocate creatine across tissue barriers and into target cells, such as neurons and myocytes. Individuals harboring mutations in the coding sequence of the human CRT1 gene develop creatine transporter deficiency (CTD), one of the pivotal underlying causes of cerebral creatine deficiency syndrome. CTD encompasses an array of clinical manifestations, including severe intellectual disability, epilepsy, autism, development delay, and motor dysfunction. CTD is characterized by the absence of cerebral creatine, which implies an indispensable role for CRT1 in supplying the brain cells with creatine. CTD-associated variants dramatically reduce or abolish creatine transport activity by CRT1. Many of these are point mutations that are known to trigger folding defects, leading to the retention of encoded CRT1 proteins in the endoplasmic reticulum and precluding their delivery to the cell surface. Misfolding of several related SLC6 transporters also gives rise to detrimental pathologic conditions in people; e.g., mutations in the dopamine transporter induce infantile parkinsonism/dystonia, while mutations in the GABA transporter 1 cause treatment-resistant epilepsy. In some cases, folding defects are amenable to rescue by small molecules, known as pharmacological and chemical chaperones, which restore the cell surface expression and transport activity of the previously non-functional proteins. Insights from the recent molecular, animal and human case studies of CTD add toward our understanding of this complex disorder and reveal the wide-ranging effects elicited upon CRT1 dysfunction. This grants novel therapeutic prospects for the treatment of patients afflicted with CTD, e.g., modifying the creatine molecule to facilitate CRT1-independent entry into brain cells, or correcting folding-deficient and loss-of-function CTD variants using pharmacochaperones and/or allosteric modulators. The latter justifies a search for additional compounds with a capacity to correct mutation-specific defects.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
