PTD-mediated delivery of α-globin chain into Κ-562 erythroleukemia cells and α-thalassemic (HBH) patients' RBCs ex vivo in the frame of Protein Replacement Therapy.
Androulla N Miliotou, Dionysia Papagiannopoulou, Efthymia Vlachaki, Martina Samiotaki, Dimitra Laspa, Stamatia Theodoridou, Asterios S Tsiftsoglou, Lefkothea C Papadopoulou
{"title":"PTD-mediated delivery of α-globin chain into Κ-562 erythroleukemia cells and α-thalassemic (HBH) patients' RBCs ex vivo in the frame of Protein Replacement Therapy.","authors":"Androulla N Miliotou, Dionysia Papagiannopoulou, Efthymia Vlachaki, Martina Samiotaki, Dimitra Laspa, Stamatia Theodoridou, Asterios S Tsiftsoglou, Lefkothea C Papadopoulou","doi":"10.1186/s40709-021-00148-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>α-Thalassemia, a congenital hemoglobinopathy, is characterized by deficiency and/or reduced levels of α-globin chains in serious forms of α-thalassemia (HbH disease/Hb Bart's). This research work deals with a Protein Replacement Therapy approach in order to manage α-thalassemia manifestations, caused by the excess of β-globin chain into HbH RBCs. The main goal was to produce the recombinant human α-globin chain in fusion with TAT, a Protein Transduction Domain, to ex vivo deliver it into HbH patients RBCs, to replace the endogenous missing α-globin chain.</p><p><strong>Results: </strong>Cloning of the α-globin coding sequence, fused to the nucleotide sequence of TAT peptide was conducted and the human recombinant fusion proteins, 10xHis-Xa<sub>SITE</sub>-α-globin-HA and 10xHis-Xa<sub>SITE</sub>-TAT-α-globin-HA were produced. The ability of human recombinant 10xHis-Xa<sub>SITE</sub>-α-globin-HA to interact in vitro with the previously produced 10xHis-Xa<sub>SITE</sub>-TAT-β-globin-HA and form α-/β-globin heterodimers, was assessed and confirmed by size exclusion chromatography. The recombinant 10xHis-Xa<sub>SITE</sub>-TAT-α-globin-HA was successfully delivered into human proerythroid K-562 cells, during the preliminary transduction evaluation experiments. Finally, the recombinant, TAT-fused α-globin was successfully transduced into RBCs, derived from HbH patients and reduced the formation of HbH-Inclusion Bodies, known to contain harmful β<sub>4</sub>-globin chain tetramers.</p><p><strong>Conclusions: </strong>Our data confirm the successful ex vivo transduction of recombinant α-globin chains in HbH RBCs to replace the missing a-globin chain and reduce the HbH-inclusion bodies, seen in α-thalassemias. These findings broaden the possibility of applying a Protein Replacement Therapy approach to module sever forms of α-thalassemia, using recombinant α-globin chains, through PTD technology.</p>","PeriodicalId":87292,"journal":{"name":"","volume":"28 1","pages":"16"},"PeriodicalIF":0.0,"publicationDate":"2021-07-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s40709-021-00148-3","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s40709-021-00148-3","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 1
Abstract
Background: α-Thalassemia, a congenital hemoglobinopathy, is characterized by deficiency and/or reduced levels of α-globin chains in serious forms of α-thalassemia (HbH disease/Hb Bart's). This research work deals with a Protein Replacement Therapy approach in order to manage α-thalassemia manifestations, caused by the excess of β-globin chain into HbH RBCs. The main goal was to produce the recombinant human α-globin chain in fusion with TAT, a Protein Transduction Domain, to ex vivo deliver it into HbH patients RBCs, to replace the endogenous missing α-globin chain.
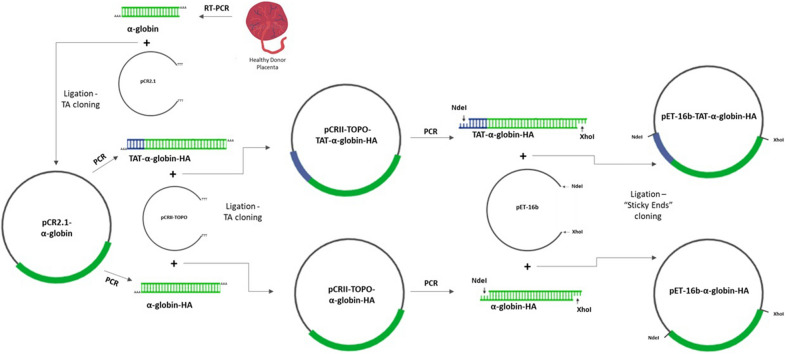
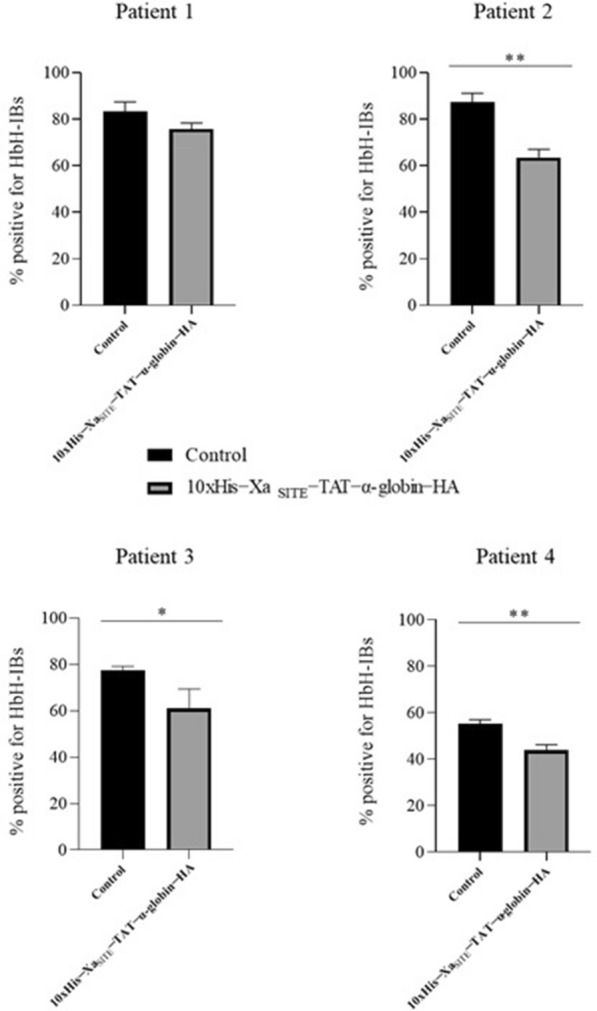
Results: Cloning of the α-globin coding sequence, fused to the nucleotide sequence of TAT peptide was conducted and the human recombinant fusion proteins, 10xHis-XaSITE-α-globin-HA and 10xHis-XaSITE-TAT-α-globin-HA were produced. The ability of human recombinant 10xHis-XaSITE-α-globin-HA to interact in vitro with the previously produced 10xHis-XaSITE-TAT-β-globin-HA and form α-/β-globin heterodimers, was assessed and confirmed by size exclusion chromatography. The recombinant 10xHis-XaSITE-TAT-α-globin-HA was successfully delivered into human proerythroid K-562 cells, during the preliminary transduction evaluation experiments. Finally, the recombinant, TAT-fused α-globin was successfully transduced into RBCs, derived from HbH patients and reduced the formation of HbH-Inclusion Bodies, known to contain harmful β4-globin chain tetramers.
Conclusions: Our data confirm the successful ex vivo transduction of recombinant α-globin chains in HbH RBCs to replace the missing a-globin chain and reduce the HbH-inclusion bodies, seen in α-thalassemias. These findings broaden the possibility of applying a Protein Replacement Therapy approach to module sever forms of α-thalassemia, using recombinant α-globin chains, through PTD technology.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
