Mohammed Alzaid , Khalid Al-Mobaireek , Mohammed Almannai , Gawahir Mukhtar , Safa Eltahir , Adnan Zafar , Abdulali P. Zada , Wadha Alotaibi
{"title":"Clinical and molecular characteristics of primary ciliary dyskinesia: A tertiary care centre experience","authors":"Mohammed Alzaid , Khalid Al-Mobaireek , Mohammed Almannai , Gawahir Mukhtar , Safa Eltahir , Adnan Zafar , Abdulali P. Zada , Wadha Alotaibi","doi":"10.1016/j.ijpam.2021.03.002","DOIUrl":null,"url":null,"abstract":"<div><h3>Background</h3><p>Primary ciliary dyskinesia (PCD) is a ciliopathy with diverse clinical and genetic findings caused by abnormal motile cilia structure and function. In this study, we describe the clinical characteristics of confirmed PCD cases in our population and report the radiological, genetic, and laboratory findings.</p></div><div><h3>Methods</h3><p>This was a retrospective, observational, single-centre study. We enrolled 18 patients who were diagnosed with confirmed PCD between 2015 and 2019. We then analyzed their data, including clinical findings and workup.</p></div><div><h3>Results</h3><p>In our cohort, 56% of patients had molecularly confirmed PCD, and RSPH9 was the most common gene identified. Transmission electron microscopy (TEM) showed an ultrastructural defect in 64% of samples, all of which matched the genetic background of the patient. Situs inversus (SI) was observed in 50% of patients, and congenital heart disease was observed in 33%. The median body mass index (BMI) was 15.87 kg/m<sup>2</sup>, with a median z score of -1.48. The median FEV1 value was 67.6% (z score - 2.43). Radiologically, bronchiectasis was noted in 81% of patients at a variable degree of severity. Lung bases were involved in 91% of patients. We were unable to correlate the genotype-phenotype findings.</p></div><div><h3>Conclusion</h3><p>We describe the clinical and molecular characteristics of patients with confirmed PCD in a tertiary centre in Saudi Arabia and report 9 new pathogenic or likely pathogenic variants in one of the PCD-associated genes.</p></div>","PeriodicalId":36646,"journal":{"name":"International Journal of Pediatrics and Adolescent Medicine","volume":"8 4","pages":"Pages 258-263"},"PeriodicalIF":0.0000,"publicationDate":"2021-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.ijpam.2021.03.002","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Pediatrics and Adolescent Medicine","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2352646721000260","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/3/11 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 4
Abstract
Background
Primary ciliary dyskinesia (PCD) is a ciliopathy with diverse clinical and genetic findings caused by abnormal motile cilia structure and function. In this study, we describe the clinical characteristics of confirmed PCD cases in our population and report the radiological, genetic, and laboratory findings.
Methods
This was a retrospective, observational, single-centre study. We enrolled 18 patients who were diagnosed with confirmed PCD between 2015 and 2019. We then analyzed their data, including clinical findings and workup.
Results
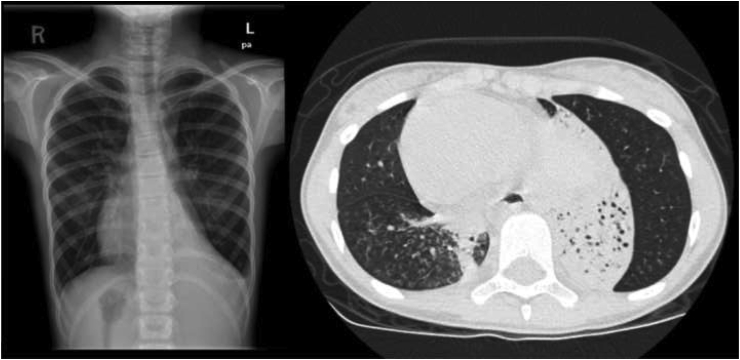
In our cohort, 56% of patients had molecularly confirmed PCD, and RSPH9 was the most common gene identified. Transmission electron microscopy (TEM) showed an ultrastructural defect in 64% of samples, all of which matched the genetic background of the patient. Situs inversus (SI) was observed in 50% of patients, and congenital heart disease was observed in 33%. The median body mass index (BMI) was 15.87 kg/m2, with a median z score of -1.48. The median FEV1 value was 67.6% (z score - 2.43). Radiologically, bronchiectasis was noted in 81% of patients at a variable degree of severity. Lung bases were involved in 91% of patients. We were unable to correlate the genotype-phenotype findings.
Conclusion
We describe the clinical and molecular characteristics of patients with confirmed PCD in a tertiary centre in Saudi Arabia and report 9 new pathogenic or likely pathogenic variants in one of the PCD-associated genes.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
