{"title":"Comparison of false-discovery rates of various decoy databases.","authors":"Sangjeong Lee, Heejin Park, Hyunwoo Kim","doi":"10.1186/s12953-021-00179-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The target-decoy strategy effectively estimates the false-discovery rate (FDR) by creating a decoy database with a size identical to that of the target database. Decoy databases are created by various methods, such as, the reverse, pseudo-reverse, shuffle, pseudo-shuffle, and the de Bruijn methods. FDR is sometimes over- or under-estimated depending on which decoy database is used because the ratios of redundant peptides in the target databases are different, that is, the numbers of unique (non-redundancy) peptides in the target and decoy databases differ.</p><p><strong>Results: </strong>We used two protein databases (the UniProt Saccharomyces cerevisiae protein database and the UniProt human protein database) to compare the FDRs of various decoy databases. When the ratio of redundant peptides in the target database is low, the FDR is not overestimated by any decoy construction method. However, if the ratio of redundant peptides in the target database is high, the FDR is overestimated when the (pseudo) shuffle decoy database is used. Additionally, human and S. cerevisiae six frame translation databases, which are large databases, also showed outcomes similar to that from the UniProt human protein database.</p><p><strong>Conclusion: </strong>The FDR must be estimated using the correction factor proposed by Elias and Gygi or that by Kim et al. when (pseudo) shuffle decoy databases are used.</p>","PeriodicalId":20857,"journal":{"name":"Proteome Science","volume":"19 1","pages":"11"},"PeriodicalIF":1.6000,"publicationDate":"2021-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8449453/pdf/","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Proteome Science","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12953-021-00179-7","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 4
Abstract
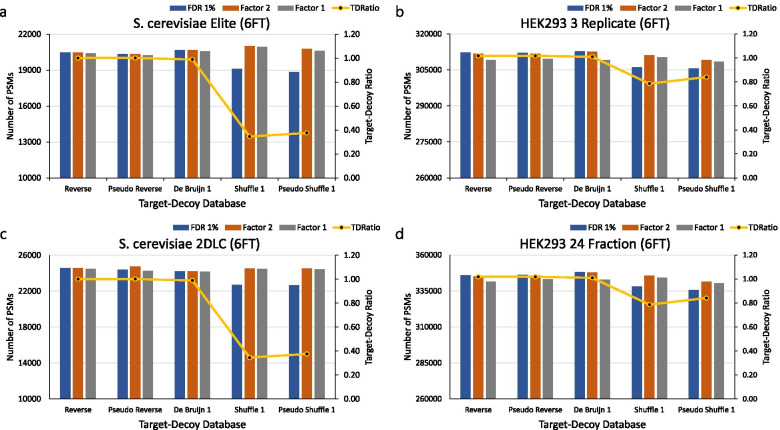
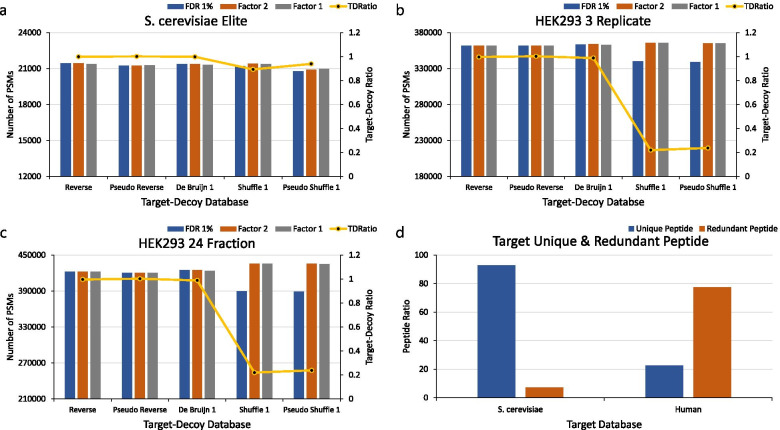
Background: The target-decoy strategy effectively estimates the false-discovery rate (FDR) by creating a decoy database with a size identical to that of the target database. Decoy databases are created by various methods, such as, the reverse, pseudo-reverse, shuffle, pseudo-shuffle, and the de Bruijn methods. FDR is sometimes over- or under-estimated depending on which decoy database is used because the ratios of redundant peptides in the target databases are different, that is, the numbers of unique (non-redundancy) peptides in the target and decoy databases differ.
Results: We used two protein databases (the UniProt Saccharomyces cerevisiae protein database and the UniProt human protein database) to compare the FDRs of various decoy databases. When the ratio of redundant peptides in the target database is low, the FDR is not overestimated by any decoy construction method. However, if the ratio of redundant peptides in the target database is high, the FDR is overestimated when the (pseudo) shuffle decoy database is used. Additionally, human and S. cerevisiae six frame translation databases, which are large databases, also showed outcomes similar to that from the UniProt human protein database.
Conclusion: The FDR must be estimated using the correction factor proposed by Elias and Gygi or that by Kim et al. when (pseudo) shuffle decoy databases are used.
期刊介绍:
Proteome Science is an open access journal publishing research in the area of systems studies. Proteome Science considers manuscripts based on all aspects of functional and structural proteomics, genomics, metabolomics, systems analysis and metabiome analysis. It encourages the submissions of studies that use large-scale or systems analysis of biomolecules in a cellular, organismal and/or environmental context.
Studies that describe novel biological or clinical insights as well as methods-focused studies that describe novel methods for the large-scale study of any and all biomolecules in cells and tissues, such as mass spectrometry, protein and nucleic acid microarrays, genomics, next-generation sequencing and computational algorithms and methods are all within the scope of Proteome Science, as are electron topography, structural methods, proteogenomics, chemical proteomics, stem cell proteomics, organelle proteomics, plant and microbial proteomics.
In spite of its name, Proteome Science considers all aspects of large-scale and systems studies because ultimately any mechanism that results in genomic and metabolomic changes will affect or be affected by the proteome. To reflect this intrinsic relationship of biological systems, Proteome Science will consider all such articles.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
