Novel mutations of the PAX6, FOXC1, and PITX2 genes cause abnormal development of the iris in Vietnamese individuals.
IF 1.4 3区 医学Q4 BIOCHEMISTRY & MOLECULAR BIOLOGYMolecular VisionPub Date : 2021-09-02eCollection Date: 2021-01-01
Ha Hai Nguyen, Chau Minh Pham, Hoa Thi Thanh Nguyen, Nhung Phuong Vu, Trang Thu Duong, Ton Dang Nguyen, Bac Duy Nguyen, Hiep Van Nguyen, Hai Van Nong
{"title":"Novel mutations of the <i>PAX6</i>, <i>FOXC1</i>, and <i>PITX2</i> genes cause abnormal development of the iris in Vietnamese individuals.","authors":"Ha Hai Nguyen, Chau Minh Pham, Hoa Thi Thanh Nguyen, Nhung Phuong Vu, Trang Thu Duong, Ton Dang Nguyen, Bac Duy Nguyen, Hiep Van Nguyen, Hai Van Nong","doi":"","DOIUrl":null,"url":null,"abstract":"<p><strong>Purpose: </strong>Congenital iris abnormality is a feature of several genetic conditions, such as aniridia syndrome and anterior segment degeneration (ASD) disorders. Aniridia syndrome is caused by mutations in the <i>PAX6</i> gene or its regulatory elements in the locus 11p13 or deletions of contiguous genes, while ASDs are the result of mutations in various genes, such as <i>PAX6</i>, <i>FOXC1</i>, <i>PITX2</i>, and <i>CYP1B1</i>. This study aims to identify pathogenic mutations in Vietnamese individuals with congenital anomalies of the iris.</p><p><strong>Methods: </strong>Genomic DNA was extracted from peripheral blood of 24 patients belonging to 15 unrelated families and their available family members. Multiplex ligation-dependent probe amplification (MLPA) was used to detect the deletions or duplications in the 11p13-14 region, including the <i>PAX6</i> gene and its neighboring genes. Direct PCR sequencing was used to screen mutations in 13 exons and flanking sequences of the <i>PAX6</i> gene. The patients without mutation in the <i>PAX6</i> locus were further analyzed with whole exome sequencing (WES). Identified mutations were tested with segregation analysis in proband family members.</p><p><strong>Results: </strong>We identified a total of 8 novel and 4 recurrent mutations in 20 of 24 affected individuals from 12 families. Among these mutations, one large deletion of the whole <i>PAX6</i> gene and another deletion of the <i>PAX6</i> downstream region containing the <i>DCDC1</i> and <i>ELP4</i> genes were identified. Eight mutations were detected in <i>PAX6</i>, including four nonsense, three frameshift, and one splice site. In addition, two point mutations were identified in the <i>FOXC1</i> and <i>PITX2</i> genes in patients without mutation in <i>PAX6</i>. Some of the mutations segregated in an autosomal dominant pattern where family members were available.</p><p><strong>Conclusions: </strong>This study provides new data on causative mutations in individuals with abnormal development of iris tissue in Vietnam. These results contribute to clinical management and genetic counseling for affected people and their families.</p>","PeriodicalId":18866,"journal":{"name":"Molecular Vision","volume":"27 ","pages":"555-563"},"PeriodicalIF":1.4000,"publicationDate":"2021-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/b2/f6/mv-v27-555.PMC8416135.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Vision","FirstCategoryId":"3","ListUrlMain":"","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Purpose: Congenital iris abnormality is a feature of several genetic conditions, such as aniridia syndrome and anterior segment degeneration (ASD) disorders. Aniridia syndrome is caused by mutations in the PAX6 gene or its regulatory elements in the locus 11p13 or deletions of contiguous genes, while ASDs are the result of mutations in various genes, such as PAX6, FOXC1, PITX2, and CYP1B1. This study aims to identify pathogenic mutations in Vietnamese individuals with congenital anomalies of the iris.
Methods: Genomic DNA was extracted from peripheral blood of 24 patients belonging to 15 unrelated families and their available family members. Multiplex ligation-dependent probe amplification (MLPA) was used to detect the deletions or duplications in the 11p13-14 region, including the PAX6 gene and its neighboring genes. Direct PCR sequencing was used to screen mutations in 13 exons and flanking sequences of the PAX6 gene. The patients without mutation in the PAX6 locus were further analyzed with whole exome sequencing (WES). Identified mutations were tested with segregation analysis in proband family members.
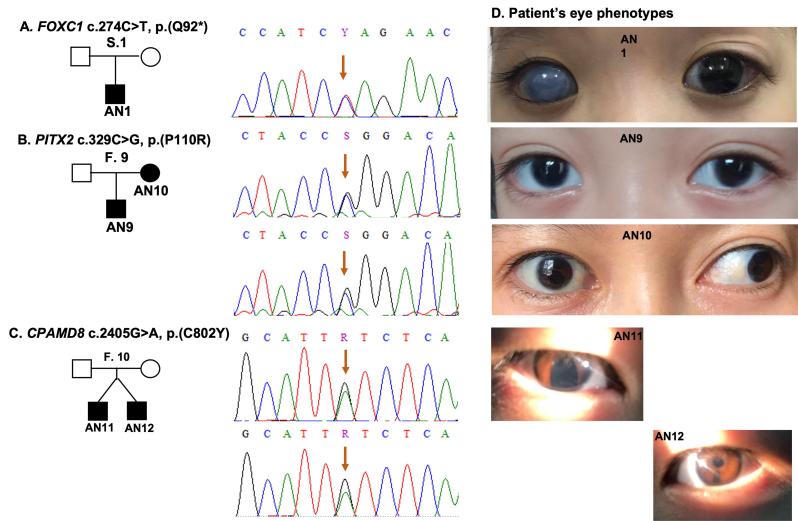
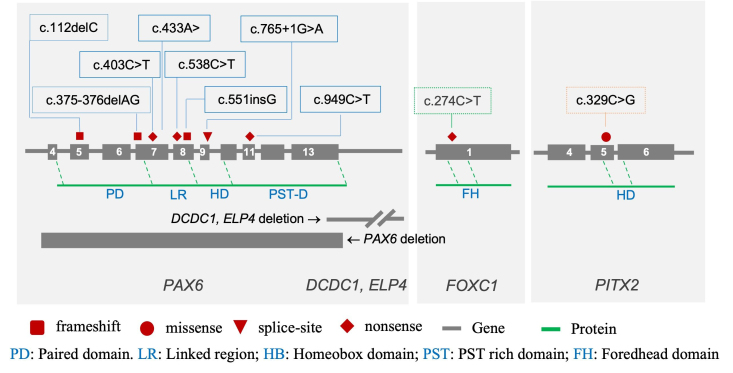
Results: We identified a total of 8 novel and 4 recurrent mutations in 20 of 24 affected individuals from 12 families. Among these mutations, one large deletion of the whole PAX6 gene and another deletion of the PAX6 downstream region containing the DCDC1 and ELP4 genes were identified. Eight mutations were detected in PAX6, including four nonsense, three frameshift, and one splice site. In addition, two point mutations were identified in the FOXC1 and PITX2 genes in patients without mutation in PAX6. Some of the mutations segregated in an autosomal dominant pattern where family members were available.
Conclusions: This study provides new data on causative mutations in individuals with abnormal development of iris tissue in Vietnam. These results contribute to clinical management and genetic counseling for affected people and their families.
期刊介绍:
Molecular Vision is a peer-reviewed journal dedicated to the dissemination of research results in molecular biology, cell biology, and the genetics of the visual system (ocular and cortical).
Molecular Vision publishes articles presenting original research that has not previously been published and comprehensive articles reviewing the current status of a particular field or topic. Submissions to Molecular Vision are subjected to rigorous peer review. Molecular Vision does NOT publish preprints.
For authors, Molecular Vision provides a rapid means of communicating important results. Access to Molecular Vision is free and unrestricted, allowing the widest possible audience for your article. Digital publishing allows you to use color images freely (and without fees). Additionally, you may publish animations, sounds, or other supplementary information that clarifies or supports your article. Each of the authors of an article may also list an electronic mail address (which will be updated upon request) to give interested readers easy access to authors.
 分享
分享



 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
