Bernard Ofoe Tetteh, Florence-Barbara Yebuah, Maame-Boatemaa Amissah-Arthur, Dzifa Dey
{"title":"Coexistence of Relapsing Polychondritis and Sickle Cell Disease in a Child.","authors":"Bernard Ofoe Tetteh, Florence-Barbara Yebuah, Maame-Boatemaa Amissah-Arthur, Dzifa Dey","doi":"10.1155/2021/3600451","DOIUrl":null,"url":null,"abstract":"<p><p>Relapsing polychondritis (RP) is a rare, severe connective tissue disease of unknown etiology affecting cartilaginous and proteoglycan-rich structures in an episodic and inflammatory manner. Approximately a third of RP cases occur in conjunction with another disease usually systemic autoimmune rheumatic disease, or myelodysplastic syndrome. Sickle cell disease (SCD) is a common inherited hematologic condition characterized by the inheritance of two abnormal hemoglobins, of which one is a hemoglobin S, presenting with severe acute and chronic complications from vaso-occlusive phenomena, which can be difficult to differentiate from RP. The pathogenesis of RP is poorly understood but suggests an autoimmune mechanism with a link to sickle cell disease yet to be established. Treatment is empiric with steroids, anti-inflammatory, and disease-modifying antirheumatic drugs being the mainstay of therapy. Severe complications occur despite treatment, with respiratory involvement being the most catastrophic. This case report reviews a complex case of RP in an 11-year-old girl with sickle cell disease (SF genotype) presenting with bilateral red painful eyes, a painful swollen left ear, and knee pain. Laboratory findings revealed elevated inflammatory markers with negative immune serology. A diagnosis of RP was made based on the patient's symptomatology, presentation, and fulfillment of 5 out of the 6 clinical features using McAdam's criteria. Management was instituted with a myriad of conventional and biologic DMARDs and other anti-inflammatory medications with no significant improvement and the development of complications of airway obstruction from disease activity and osteoporotic fracture from steroid therapy and underlying hemoglobinopathy. In children, the diagnosis of RP is delayed or overlooked due to its low incidence, variability in clinical symptoms, or sharing similar clinical features with other coexisting disease entities. This article reports its occurrence in the pediatric population and highlights the difficulty in managing such cases as there are no defined standard treatment protocols.</p>","PeriodicalId":9622,"journal":{"name":"Case Reports in Rheumatology","volume":"2021 ","pages":"3600451"},"PeriodicalIF":0.0000,"publicationDate":"2021-11-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8635928/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Rheumatology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2021/3600451","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
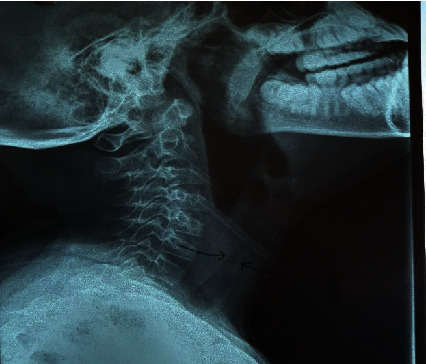
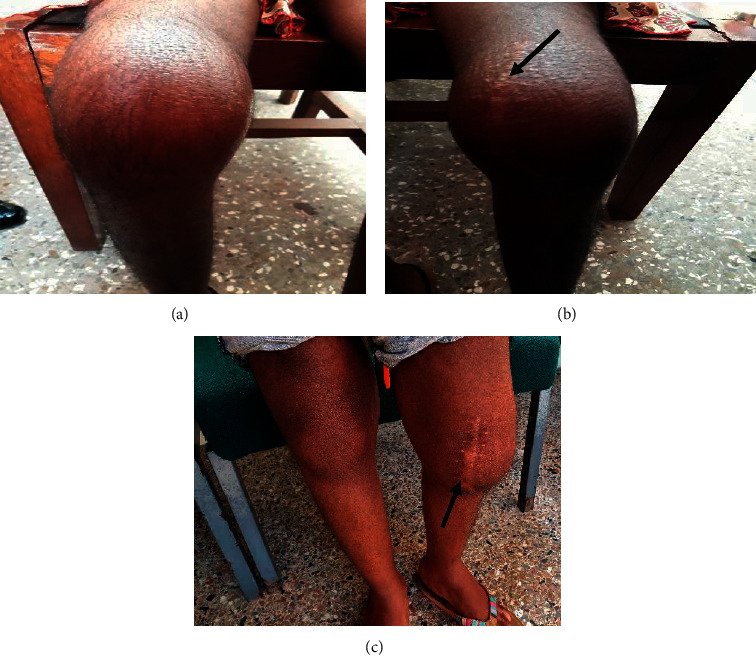
Relapsing polychondritis (RP) is a rare, severe connective tissue disease of unknown etiology affecting cartilaginous and proteoglycan-rich structures in an episodic and inflammatory manner. Approximately a third of RP cases occur in conjunction with another disease usually systemic autoimmune rheumatic disease, or myelodysplastic syndrome. Sickle cell disease (SCD) is a common inherited hematologic condition characterized by the inheritance of two abnormal hemoglobins, of which one is a hemoglobin S, presenting with severe acute and chronic complications from vaso-occlusive phenomena, which can be difficult to differentiate from RP. The pathogenesis of RP is poorly understood but suggests an autoimmune mechanism with a link to sickle cell disease yet to be established. Treatment is empiric with steroids, anti-inflammatory, and disease-modifying antirheumatic drugs being the mainstay of therapy. Severe complications occur despite treatment, with respiratory involvement being the most catastrophic. This case report reviews a complex case of RP in an 11-year-old girl with sickle cell disease (SF genotype) presenting with bilateral red painful eyes, a painful swollen left ear, and knee pain. Laboratory findings revealed elevated inflammatory markers with negative immune serology. A diagnosis of RP was made based on the patient's symptomatology, presentation, and fulfillment of 5 out of the 6 clinical features using McAdam's criteria. Management was instituted with a myriad of conventional and biologic DMARDs and other anti-inflammatory medications with no significant improvement and the development of complications of airway obstruction from disease activity and osteoporotic fracture from steroid therapy and underlying hemoglobinopathy. In children, the diagnosis of RP is delayed or overlooked due to its low incidence, variability in clinical symptoms, or sharing similar clinical features with other coexisting disease entities. This article reports its occurrence in the pediatric population and highlights the difficulty in managing such cases as there are no defined standard treatment protocols.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
