Kazuhiro Aoki , Adam D. Heaps , Kevin A. Strauss , Michael Tiemeyer
{"title":"Mass spectrometric quantification of plasma glycosphingolipids in human GM3 ganglioside deficiency","authors":"Kazuhiro Aoki , Adam D. Heaps , Kevin A. Strauss , Michael Tiemeyer","doi":"10.1016/j.clinms.2019.03.001","DOIUrl":null,"url":null,"abstract":"<div><h3>Background</h3><p>Among Amish communities of North America, biallelic mutations of <em>ST3GAL5</em> (c.694C > T) eliminate synthesis of GM3 and its derivative downstream a- and b-series gangliosides. Systemic ganglioside deficiency is associated with infantile onset psychomotor retardation, slow brain growth, intractable epilepsy, deafness, and cortical visual impairment. We developed a robust quantitative assay to simultaneously characterize glycan and ceramide moieties of plasma glycosphingolipids (GSLs) among <em>ST3GAL5</em> c.694C > T homozygotes (n = 8), their heterozygous siblings (n = 24), and wild type control (n = 19) individuals.</p></div><div><h3>Methods</h3><p>Following extraction and saponification of total plasma lipids, GSLs were purified on a tC18 cartridge column, permethylated, and subjected to nanospray ionization mass spectrometry utilizing neutral loss scanning and data-dependent acquisition. Plasma GSLs were quantified against appropriate synthetic standards.</p></div><div><h3>Results</h3><p>Our method demonstrated linearity from 5 to 250 μl of plasma. Recovery of synthetic GSLs spiked into plasma was 99–104% with no matrix interference. Quantitative plasma GSL profiles discriminated among <em>ST3GAL5</em> genotypes: GM3 and GD3 were undetectable in <em>ST3GAL5</em> c.694C > T homozygotes, who had markedly elevated lactosylceramide (19.17 ± 4.20 nmol/ml) relative to heterozygous siblings (9.62 ± 2.46 nmol/ml) and wild type controls (6.55 ± 2.16 nmol/ml). Children with systemic ganglioside deficiency had a distinctive shift in ceramide composition toward higher mass species.</p></div><div><h3>Conclusions</h3><p>Our quantitative glycolipidomics method discriminates among <em>ST3GAL5</em> c.694C > T genotypes, can reveal subtle structural heterogeneity, and represents a useful new strategy to diagnose and monitor GSL disorders in humans.</p></div>","PeriodicalId":72613,"journal":{"name":"","volume":"14 ","pages":"Pages 106-114"},"PeriodicalIF":0.0,"publicationDate":"2019-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.clinms.2019.03.001","citationCount":"9","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2376999818300564","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2019/3/16 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 9
Abstract
Background
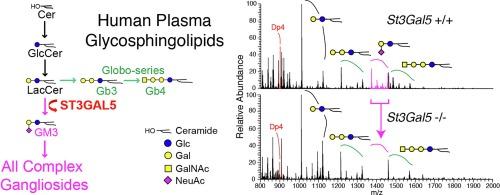
Among Amish communities of North America, biallelic mutations of ST3GAL5 (c.694C > T) eliminate synthesis of GM3 and its derivative downstream a- and b-series gangliosides. Systemic ganglioside deficiency is associated with infantile onset psychomotor retardation, slow brain growth, intractable epilepsy, deafness, and cortical visual impairment. We developed a robust quantitative assay to simultaneously characterize glycan and ceramide moieties of plasma glycosphingolipids (GSLs) among ST3GAL5 c.694C > T homozygotes (n = 8), their heterozygous siblings (n = 24), and wild type control (n = 19) individuals.
Methods
Following extraction and saponification of total plasma lipids, GSLs were purified on a tC18 cartridge column, permethylated, and subjected to nanospray ionization mass spectrometry utilizing neutral loss scanning and data-dependent acquisition. Plasma GSLs were quantified against appropriate synthetic standards.
Results
Our method demonstrated linearity from 5 to 250 μl of plasma. Recovery of synthetic GSLs spiked into plasma was 99–104% with no matrix interference. Quantitative plasma GSL profiles discriminated among ST3GAL5 genotypes: GM3 and GD3 were undetectable in ST3GAL5 c.694C > T homozygotes, who had markedly elevated lactosylceramide (19.17 ± 4.20 nmol/ml) relative to heterozygous siblings (9.62 ± 2.46 nmol/ml) and wild type controls (6.55 ± 2.16 nmol/ml). Children with systemic ganglioside deficiency had a distinctive shift in ceramide composition toward higher mass species.
Conclusions
Our quantitative glycolipidomics method discriminates among ST3GAL5 c.694C > T genotypes, can reveal subtle structural heterogeneity, and represents a useful new strategy to diagnose and monitor GSL disorders in humans.


 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
