Osman Mutluhan Uğurel, Oğuz Ata, Dilek Turgut-Balik
{"title":"Genomic chronicle of SARS-CoV-2: a mutational analysis with over 1 million genome sequences.","authors":"Osman Mutluhan Uğurel, Oğuz Ata, Dilek Turgut-Balik","doi":"10.3906/biy-2106-8","DOIUrl":null,"url":null,"abstract":"<p><p>Use of information technologies to analyse big data on SARS-CoV-2 genome provides an insight for tracking variations and examining the evolution of the virus. Nevertheless, storing, processing, alignment and analyses of these numerous genomes are still a challenge. In this study, over 1 million SARS-CoV-2 genomes have been analysed to show distribution and relationship of variations that could enlighten development and evolution of the virus. In all genomes analysed in this study, a total of over 215M SNVs have been detected and average number of SNV per isolate was found to be 21.83. Single nucleotide variant (SNV) average is observed to reach 31.25 just in March 2021. The average variation number of isolates is increasing and compromising with total case numbers around the world. Remarkably, cytosine deamination, which is one of the most important biochemical processes in the evolutionary development of coronaviruses, accounts for 46% of all SNVs seen in SARS-CoV-2 genomes within 16 months. This study is one of the most comprehensive SARS-CoV-2 genomic analysis study in terms of number of genomes analysed in an academic publication so far, and reported results could be useful in monitoring the development of SARS-CoV-2.</p>","PeriodicalId":23375,"journal":{"name":"Turkish journal of biology = Turk biyoloji dergisi","volume":"45 4","pages":"425-435"},"PeriodicalIF":0.0000,"publicationDate":"2021-08-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8573839/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Turkish journal of biology = Turk biyoloji dergisi","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3906/biy-2106-8","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
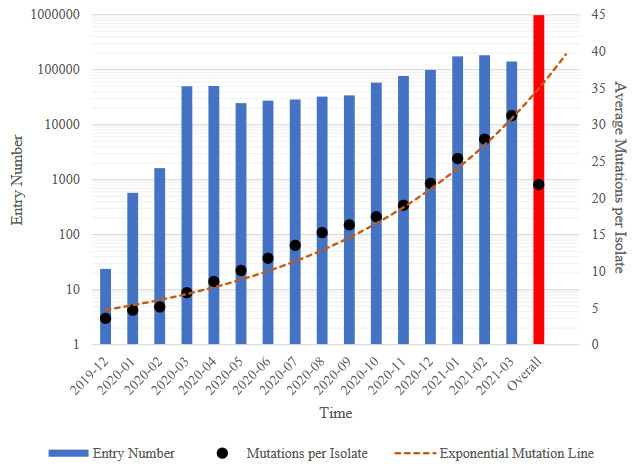
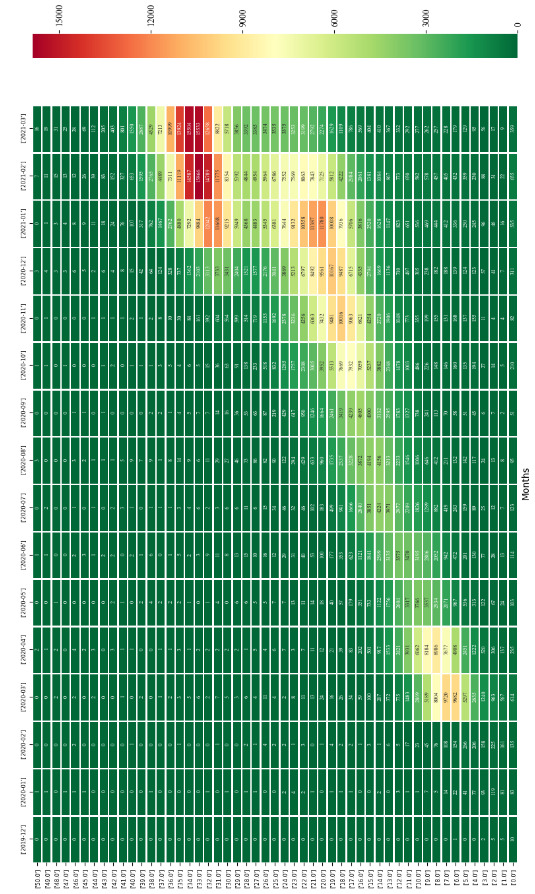
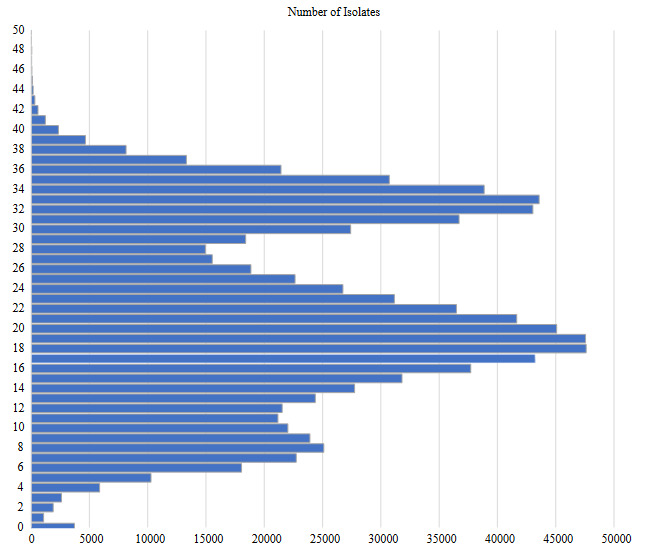
Use of information technologies to analyse big data on SARS-CoV-2 genome provides an insight for tracking variations and examining the evolution of the virus. Nevertheless, storing, processing, alignment and analyses of these numerous genomes are still a challenge. In this study, over 1 million SARS-CoV-2 genomes have been analysed to show distribution and relationship of variations that could enlighten development and evolution of the virus. In all genomes analysed in this study, a total of over 215M SNVs have been detected and average number of SNV per isolate was found to be 21.83. Single nucleotide variant (SNV) average is observed to reach 31.25 just in March 2021. The average variation number of isolates is increasing and compromising with total case numbers around the world. Remarkably, cytosine deamination, which is one of the most important biochemical processes in the evolutionary development of coronaviruses, accounts for 46% of all SNVs seen in SARS-CoV-2 genomes within 16 months. This study is one of the most comprehensive SARS-CoV-2 genomic analysis study in terms of number of genomes analysed in an academic publication so far, and reported results could be useful in monitoring the development of SARS-CoV-2.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
