Elaine Yuen Ling Au, Edmund Kwok Kwan Tung, Ricky Wai Ki Ip, Philip Hei Li
{"title":"Novel <i>MAGT1</i> Mutation Found in the First Chinese XMEN in Hong Kong.","authors":"Elaine Yuen Ling Au, Edmund Kwok Kwan Tung, Ricky Wai Ki Ip, Philip Hei Li","doi":"10.1155/2022/2390167","DOIUrl":null,"url":null,"abstract":"<p><p>The availability of next-generation sequencing (NGS) helps to resolve many of the diagnostic odysseys. Common variable immunodeficiency disease (CVID) is an entity encompassing a heterogenous group of conditions with hypogammaglobulinemia, and it is a diagnosis of exclusion. In recent years, with the advances of molecular diagnostics, more and more patients have been reclassified with more defined entities after their genetic causes were found. Here, we reported a young man, who was managed as CVID since childhood, presenting with recurrent infection, hypogammaglobulinemia, and immune thrombocytopenia (ITP). Finally, more than a decade after initial presentation, gene panel testing revealed a novel mutation in the MAGT1 gene. Collectively, the genetic findings and clinical presentations confirm the diagnosis of X-linked immunodeficiency with magnesium defect and Epstein-Barr virus infection and neoplasia (XMEN). MAGT1 is an evolutionarily conserved, magnesium-specific transporter expressed in all mammalian cells that plays an essential role in magnesium homeostasis. MAGT1 also acts as an accessory protein for STT3B, as catalytic subunits of the oligosaccharyltransferase protein complex, which carries out glycan chain transfer to proteins in the endoplasmic reticulum during N-glycosylation. Glycans play an essential role in the stability, maturation, and localization in glycoproteins that are important in our immune cells' function. Mutation of the gene resulted in a rare X-linked recessive condition XMEN. The disease has complete penetrance but variable expressivity. It is mainly associated with immunodeficiency, immunodysregulation, and predisposition to EBV-associated lymphoproliferation. Extraimmune manifestations have also been reported in some patient cohorts, including hepatic and neurological abnormalities. Overall, the presentation varies among patients and overlaps with other clinical entities, in which diagnosis is challenging. Before the era of NGS, traditional workup hinges heavily on phenotype studies, followed by single-gene sequencing. The diagnostic yield is low, and a significant delay in diagnosis is common. This case illustrated the importance of early consideration of molecular studies in complex immunological cases without obvious secondary causes as an integral part of patient management.</p>","PeriodicalId":42865,"journal":{"name":"Case Reports in Immunology","volume":"2022 ","pages":"2390167"},"PeriodicalIF":1.5000,"publicationDate":"2022-02-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8860550/pdf/","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Immunology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2022/2390167","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
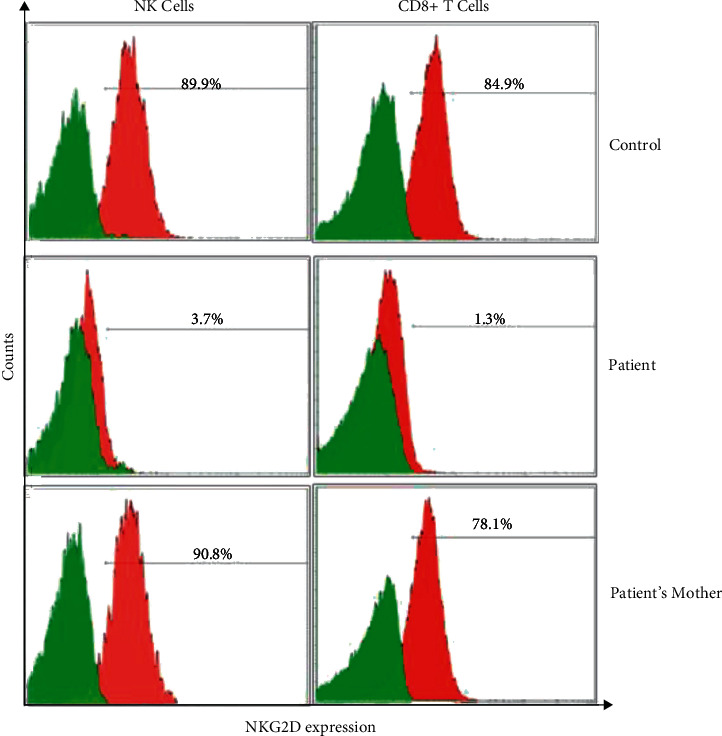
The availability of next-generation sequencing (NGS) helps to resolve many of the diagnostic odysseys. Common variable immunodeficiency disease (CVID) is an entity encompassing a heterogenous group of conditions with hypogammaglobulinemia, and it is a diagnosis of exclusion. In recent years, with the advances of molecular diagnostics, more and more patients have been reclassified with more defined entities after their genetic causes were found. Here, we reported a young man, who was managed as CVID since childhood, presenting with recurrent infection, hypogammaglobulinemia, and immune thrombocytopenia (ITP). Finally, more than a decade after initial presentation, gene panel testing revealed a novel mutation in the MAGT1 gene. Collectively, the genetic findings and clinical presentations confirm the diagnosis of X-linked immunodeficiency with magnesium defect and Epstein-Barr virus infection and neoplasia (XMEN). MAGT1 is an evolutionarily conserved, magnesium-specific transporter expressed in all mammalian cells that plays an essential role in magnesium homeostasis. MAGT1 also acts as an accessory protein for STT3B, as catalytic subunits of the oligosaccharyltransferase protein complex, which carries out glycan chain transfer to proteins in the endoplasmic reticulum during N-glycosylation. Glycans play an essential role in the stability, maturation, and localization in glycoproteins that are important in our immune cells' function. Mutation of the gene resulted in a rare X-linked recessive condition XMEN. The disease has complete penetrance but variable expressivity. It is mainly associated with immunodeficiency, immunodysregulation, and predisposition to EBV-associated lymphoproliferation. Extraimmune manifestations have also been reported in some patient cohorts, including hepatic and neurological abnormalities. Overall, the presentation varies among patients and overlaps with other clinical entities, in which diagnosis is challenging. Before the era of NGS, traditional workup hinges heavily on phenotype studies, followed by single-gene sequencing. The diagnostic yield is low, and a significant delay in diagnosis is common. This case illustrated the importance of early consideration of molecular studies in complex immunological cases without obvious secondary causes as an integral part of patient management.
期刊介绍:
Case Reports in Immunology is a peer-reviewed, Open Access journal that publishes case reports and case series related to allergies, immunodeficiencies, autoimmune diseases, immune disorders, cancer immunology and transplantation immunology.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
