{"title":"Partial nephrogenic diabetes insipidus with a novel arginine vasopressin receptor 2 gene variant.","authors":"Atsushi Ishida, Haruo Mizuno, Kohei Aoyama, Shiori Sasaki, Yutaka Negishi, Takeshi Arakawa, Takayasu Mori","doi":"10.1297/cpe.2021-0029","DOIUrl":null,"url":null,"abstract":"<p><p>X-linked nephrogenic diabetes insipidus (NDI) is caused by variations in arginine vasopressin receptor 2 (<i>AVPR2</i>). Some patients show partial resistance to arginine vasopressin (AVP). A 19-month-old Japanese boy presented with polydipsia since infancy. His mother had a history of polydipsia during pregnancy, and his maternal granduncle also had polydipsia. Intermediate urine osmolality and markedly high plasma AVP levels were observed in the water deprivation test. Subsequent pitressin administration caused no further elevation in urine osmolality. We diagnosed the patient with partial NDI, initiated therapy with hydrochlorothiazide, and placed him on a low-sodium diet. Although his urine volume decreased by 20-30% after the initiation of therapy, progressive hydronephrosis and growth retardation developed 2 years later. We investigated his genetic background by multiplex targeted sequencing of genes associated with inherited renal diseases, including <i>AVPR2</i> and aquaporin-2 (<i>AQP2</i>). We identified a hemizygous missense variant in <i>AVPR2</i> NM_000054:c.371A>G,p.(Tyr124Cys) in the boy and a heterozygous variant in the mother at the same locus. Distinguishing partial NDI from primary polydipsia is difficult because of its mild symptoms. Markedly elevated plasma AVP levels with intermediate urine osmolality may suggest partial NDI, and genetic analysis can be useful for such patients.</p>","PeriodicalId":10678,"journal":{"name":"Clinical Pediatric Endocrinology","volume":"31 1","pages":"44-49"},"PeriodicalIF":1.2000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/19/88/cpe-31-044.PMC8713061.pdf","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pediatric Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1297/cpe.2021-0029","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/11/1 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 1
Abstract
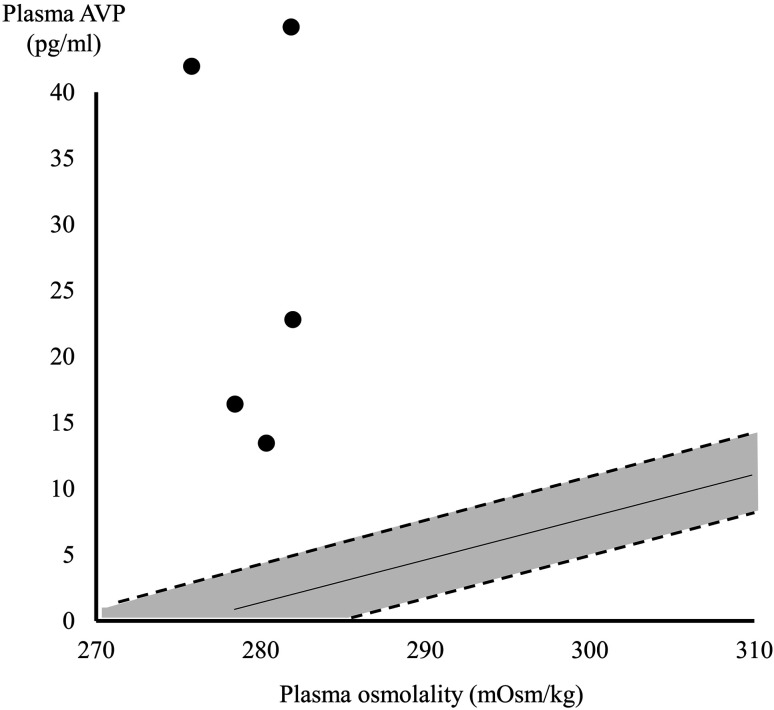
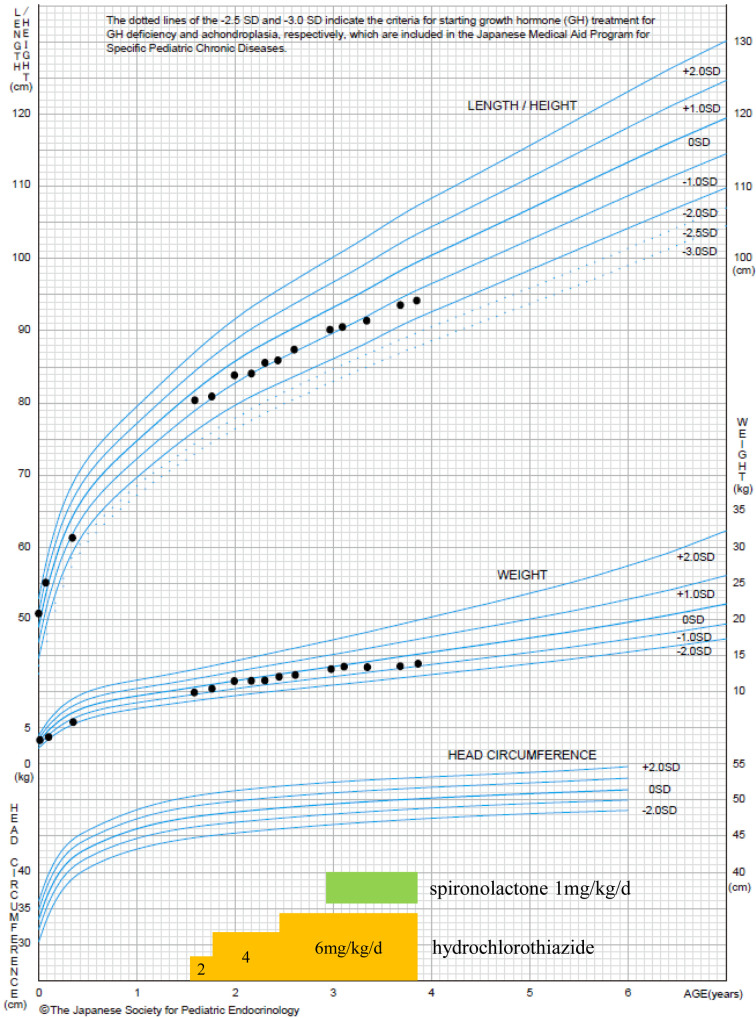
X-linked nephrogenic diabetes insipidus (NDI) is caused by variations in arginine vasopressin receptor 2 (AVPR2). Some patients show partial resistance to arginine vasopressin (AVP). A 19-month-old Japanese boy presented with polydipsia since infancy. His mother had a history of polydipsia during pregnancy, and his maternal granduncle also had polydipsia. Intermediate urine osmolality and markedly high plasma AVP levels were observed in the water deprivation test. Subsequent pitressin administration caused no further elevation in urine osmolality. We diagnosed the patient with partial NDI, initiated therapy with hydrochlorothiazide, and placed him on a low-sodium diet. Although his urine volume decreased by 20-30% after the initiation of therapy, progressive hydronephrosis and growth retardation developed 2 years later. We investigated his genetic background by multiplex targeted sequencing of genes associated with inherited renal diseases, including AVPR2 and aquaporin-2 (AQP2). We identified a hemizygous missense variant in AVPR2 NM_000054:c.371A>G,p.(Tyr124Cys) in the boy and a heterozygous variant in the mother at the same locus. Distinguishing partial NDI from primary polydipsia is difficult because of its mild symptoms. Markedly elevated plasma AVP levels with intermediate urine osmolality may suggest partial NDI, and genetic analysis can be useful for such patients.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
