{"title":"Statistical models and computational tools for predicting complex traits and diseases.","authors":"Wonil Chung","doi":"10.5808/gi.21053","DOIUrl":null,"url":null,"abstract":"<p><p>Predicting individual traits and diseases from genetic variants is critical to fulfilling the promise of personalized medicine. The genetic variants from genome-wide association studies (GWAS), including variants well below GWAS significance, can be aggregated into highly significant predictions across a wide range of complex traits and diseases. The recent arrival of large-sample public biobanks enables highly accurate polygenic predictions based on genetic variants across the whole genome. Various statistical methodologies and diverse computational tools have been introduced and developed to computed the polygenic risk score (PRS) more accurately. However, many researchers utilize PRS tools without a thorough understanding of the underlying model and how to specify the parameters for the best performance. It is advantageous to study the statistical models implemented in computational tools for PRS estimation and the formulas of parameters to be specified. Here, we review a variety of recent statistical methodologies and computational tools for PRS computation.</p>","PeriodicalId":36591,"journal":{"name":"Genomics and Informatics","volume":"19 4","pages":"e36"},"PeriodicalIF":0.0000,"publicationDate":"2021-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8752975/pdf/","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genomics and Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5808/gi.21053","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/12/31 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
引用次数: 4
Abstract
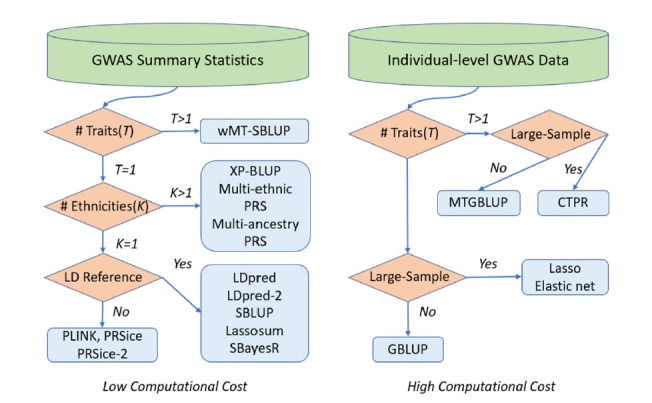
Predicting individual traits and diseases from genetic variants is critical to fulfilling the promise of personalized medicine. The genetic variants from genome-wide association studies (GWAS), including variants well below GWAS significance, can be aggregated into highly significant predictions across a wide range of complex traits and diseases. The recent arrival of large-sample public biobanks enables highly accurate polygenic predictions based on genetic variants across the whole genome. Various statistical methodologies and diverse computational tools have been introduced and developed to computed the polygenic risk score (PRS) more accurately. However, many researchers utilize PRS tools without a thorough understanding of the underlying model and how to specify the parameters for the best performance. It is advantageous to study the statistical models implemented in computational tools for PRS estimation and the formulas of parameters to be specified. Here, we review a variety of recent statistical methodologies and computational tools for PRS computation.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
