{"title":"Caveolin-1 Scaffolding Domain Peptide Regulates Colon Endothelial Cell Survival through JNK Pathway.","authors":"Kai Fang, Christopher G Kevil","doi":"10.1155/2020/6150942","DOIUrl":null,"url":null,"abstract":"<p><p>It has been reported that pathological angiogenesis contributes to both experimental colitis and inflammatory bowel disease. Recently, we demonstrated that endothelial caveolin-1 plays a key role in the pathological angiogenesis of dextran sodium sulfate (DSS) colitis. However, the molecular mechanism of caveolin-1 regulation of endothelial function is unknown. In this study, we examined how the antennapedia- (AP-) conjugated caveolin-1 scaffolding domain (AP-Cav) modulates vascular endothelial growth factor- (VEGF-) dependent colon endothelial cell angiogenic responses, as seen during colitis. We used mouse colon endothelial cells and found that AP-Cav significantly inhibited VEGF-mediated bromodeoxyuridine (BrdU) incorporation into colon microvascular endothelial cells. AP-Cav significantly blunted VEGF-dependent extracellular signal-regulated kinase 1/2 (ERK 1/2) phosphorylation at 10 minutes and 2 hours after stimulation, compared with the AP control peptide. AP-Cav + VEGF-A treatment also significantly increased c-Jun N-terminal kinase (JNK) phosphorylation at 2 hours. AP-Cav + VEGF-A treatment significantly downregulated retinoblastoma (Rb) protein levels, upregulated cleaved caspase-3 protein levels at 4 hours, and induced apoptosis. Thus, our study suggests that disruption of endothelial caveolin-1 function via the AP-Cav diverts VEGF signaling responses away from endothelial cell proliferation and toward apoptosis through the inhibition of mitogen-activated protein (MAP) kinase signaling and the induction of JNK-associated apoptosis.</p>","PeriodicalId":14004,"journal":{"name":"International Journal of Inflammation","volume":"2020 ","pages":"6150942"},"PeriodicalIF":2.0000,"publicationDate":"2020-03-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2020/6150942","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Inflammation","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2020/6150942","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
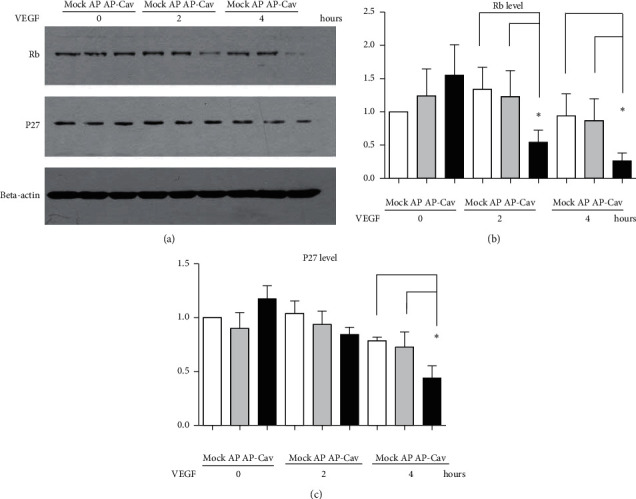
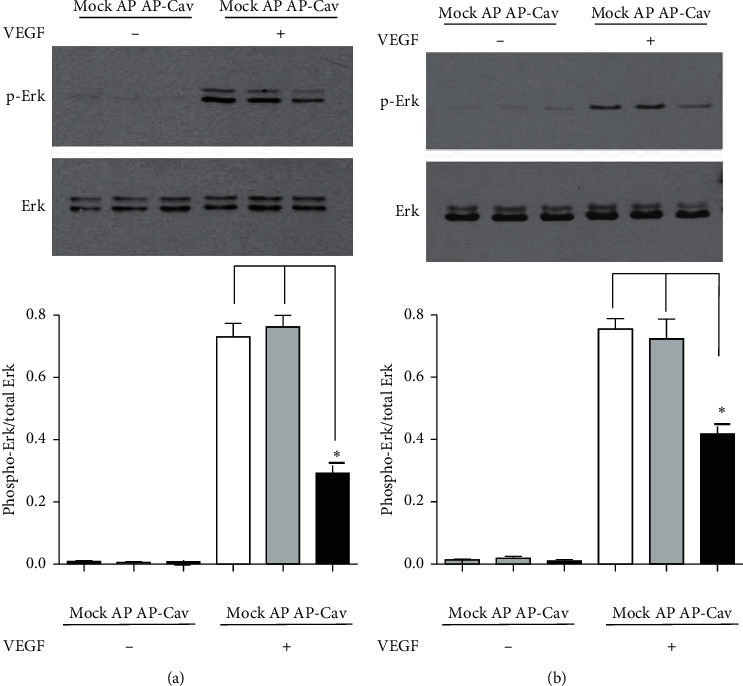
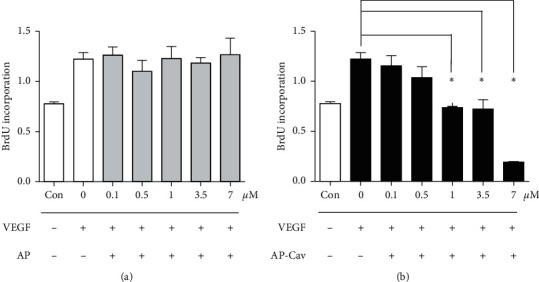
It has been reported that pathological angiogenesis contributes to both experimental colitis and inflammatory bowel disease. Recently, we demonstrated that endothelial caveolin-1 plays a key role in the pathological angiogenesis of dextran sodium sulfate (DSS) colitis. However, the molecular mechanism of caveolin-1 regulation of endothelial function is unknown. In this study, we examined how the antennapedia- (AP-) conjugated caveolin-1 scaffolding domain (AP-Cav) modulates vascular endothelial growth factor- (VEGF-) dependent colon endothelial cell angiogenic responses, as seen during colitis. We used mouse colon endothelial cells and found that AP-Cav significantly inhibited VEGF-mediated bromodeoxyuridine (BrdU) incorporation into colon microvascular endothelial cells. AP-Cav significantly blunted VEGF-dependent extracellular signal-regulated kinase 1/2 (ERK 1/2) phosphorylation at 10 minutes and 2 hours after stimulation, compared with the AP control peptide. AP-Cav + VEGF-A treatment also significantly increased c-Jun N-terminal kinase (JNK) phosphorylation at 2 hours. AP-Cav + VEGF-A treatment significantly downregulated retinoblastoma (Rb) protein levels, upregulated cleaved caspase-3 protein levels at 4 hours, and induced apoptosis. Thus, our study suggests that disruption of endothelial caveolin-1 function via the AP-Cav diverts VEGF signaling responses away from endothelial cell proliferation and toward apoptosis through the inhibition of mitogen-activated protein (MAP) kinase signaling and the induction of JNK-associated apoptosis.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
