Clara Pardinhas, Gustavo Santo, Luís Escada, Jorge Rodrigues, Maria Rosário Almeida, Rui Alves, Manuel Salgado
{"title":"A Case of Deficiency of Adenosine Deaminase 2: 28 years of Diagnostic Challenges.","authors":"Clara Pardinhas, Gustavo Santo, Luís Escada, Jorge Rodrigues, Maria Rosário Almeida, Rui Alves, Manuel Salgado","doi":"10.1159/000517141","DOIUrl":null,"url":null,"abstract":"<p><p>Deficiency of adenosine deaminase 2 (DADA2) is a unique monogenic autoinflammatory disease caused by autosomal recessive loss-of-function mutations in the CECR1 gene which presents as childhood-onset small- and medium-vessel vasculitis. Previously, many of these patients were misdiagnosed and thought to have clinical features of systemic polyarteritis nodosum, which negatively influenced its outcome, since TNF inhibitors seem to have efficacy on the vasculitic phenotype of DADA2. We present a case of a 28-year-old woman with a lifelong unknown syndrome and unique clinical manifestations recently recognized as DADA2. The first manifestation, at 3 months of age, was an episode of facial paralysis during which renovascular hypertension was diagnosed. Later, she developed episodes of prolonged fever, polyarthritis, Raynaud's phenomenon, gastrointestinal bleeding, and intracerebral hemorrhage. This inflammatory state ultimately led to the development of amyloid A amyloidosis and renal insufficiency.</p>","PeriodicalId":9599,"journal":{"name":"Case Reports in Nephrology and Dialysis","volume":"11 3","pages":"340-347"},"PeriodicalIF":0.9000,"publicationDate":"2021-11-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/93/cc/cnd-0011-0340.PMC8739640.pdf","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Nephrology and Dialysis","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000517141","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/9/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"UROLOGY & NEPHROLOGY","Score":null,"Total":0}
引用次数: 3
Abstract
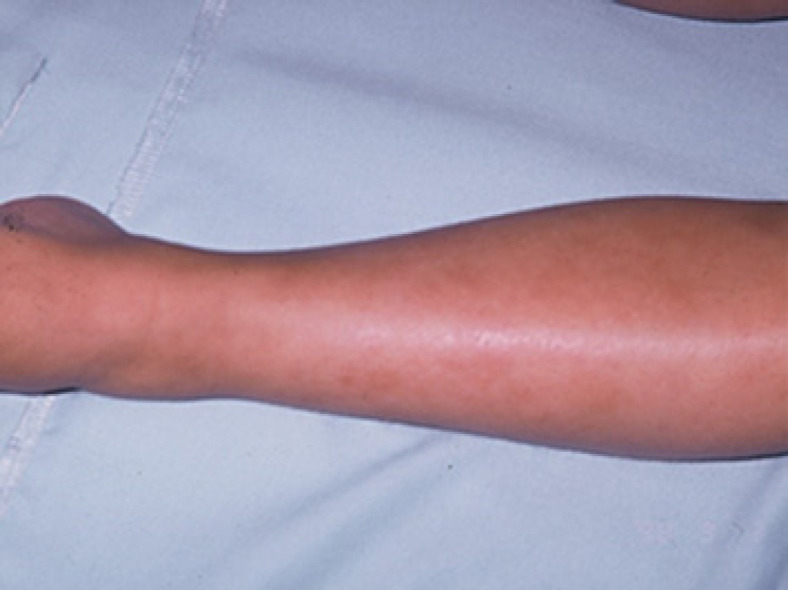
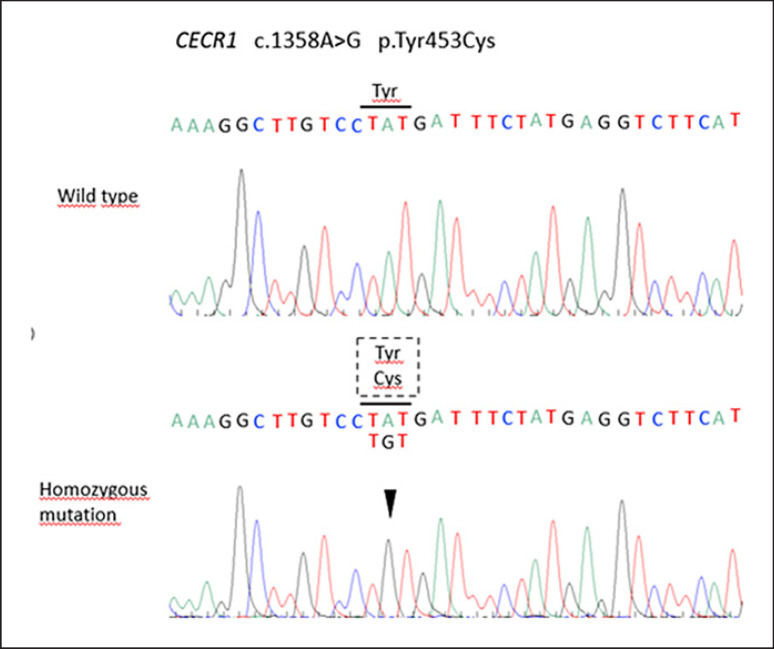
Deficiency of adenosine deaminase 2 (DADA2) is a unique monogenic autoinflammatory disease caused by autosomal recessive loss-of-function mutations in the CECR1 gene which presents as childhood-onset small- and medium-vessel vasculitis. Previously, many of these patients were misdiagnosed and thought to have clinical features of systemic polyarteritis nodosum, which negatively influenced its outcome, since TNF inhibitors seem to have efficacy on the vasculitic phenotype of DADA2. We present a case of a 28-year-old woman with a lifelong unknown syndrome and unique clinical manifestations recently recognized as DADA2. The first manifestation, at 3 months of age, was an episode of facial paralysis during which renovascular hypertension was diagnosed. Later, she developed episodes of prolonged fever, polyarthritis, Raynaud's phenomenon, gastrointestinal bleeding, and intracerebral hemorrhage. This inflammatory state ultimately led to the development of amyloid A amyloidosis and renal insufficiency.
期刊介绍:
This peer-reviewed online-only journal publishes original case reports covering the entire spectrum of nephrology and dialysis, including genetic susceptibility, clinical presentation, diagnosis, treatment or prevention, toxicities of therapy, critical care, supportive care, quality-of-life and survival issues. The journal will also accept case reports dealing with the use of novel technologies, both in the arena of diagnosis and treatment. Supplementary material is welcomed.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
