Agnesa Panferova, Kseniya Yu Sinichenkova, Meriam Abu Jabal, Natalia Usman, Anastasya Sharlai, Vitalii Roshchin, Dmitry Konovalov, Alexander Druy
{"title":"EWSR1-TFCP2 in an adolescent represents an extremely rare and aggressive form of intraosseous spindle cell rhabdomyosarcomas.","authors":"Agnesa Panferova, Kseniya Yu Sinichenkova, Meriam Abu Jabal, Natalia Usman, Anastasya Sharlai, Vitalii Roshchin, Dmitry Konovalov, Alexander Druy","doi":"10.1101/mcs.a006209","DOIUrl":null,"url":null,"abstract":"<p><p>The WHO Classification of Tumors of Soft Tissue and Bone subdivides rhabdomyosarcomas (RMS) into alveolar, embryonal, pleomorphic, and spindle cell RMS. Advances in molecular genetic diagnostics have made it possible to identify new RMS subgroups within traditional morphological entities. One of these subgroups comprises rare tumors characterized by epithelioid and spindle cell morphology, highly aggressive clinical course with pronounced tendency to intraosseous growth, and the presence of pathognomonic recurring genetic aberrations- chimeric genes/transcripts EWSR1::TFCP2, FUS::TFCP2, or MEIS1::NCOA2. Starting from 2018, only 26 reported cases of RMS have been assigned to this subgroup. The rarity of such tumors hampers their correct diagnostics for both anatomic pathologists and molecular oncologists. Here we describe a clinical case of intraosseous spindle cell RMS expressing EWSR1::TFCP2 fusion gene, encountered for the first time in our practice, in a 16-year-old female patient presenting with mandibular lesion. The diagnostic process took considerable time and involved RNA sequencing; a high-throughput method of molecular genetic research. The tumor was extremely aggressive, showing resistance to polychemotherapy, radiation therapy, and crizotinib targeted therapy, with the fatal outcome.</p>","PeriodicalId":10360,"journal":{"name":"Cold Spring Harbor Molecular Case Studies","volume":" ","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2022-06-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/27/02/MCS006209Pan.PMC9528966.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cold Spring Harbor Molecular Case Studies","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/mcs.a006209","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
Abstract
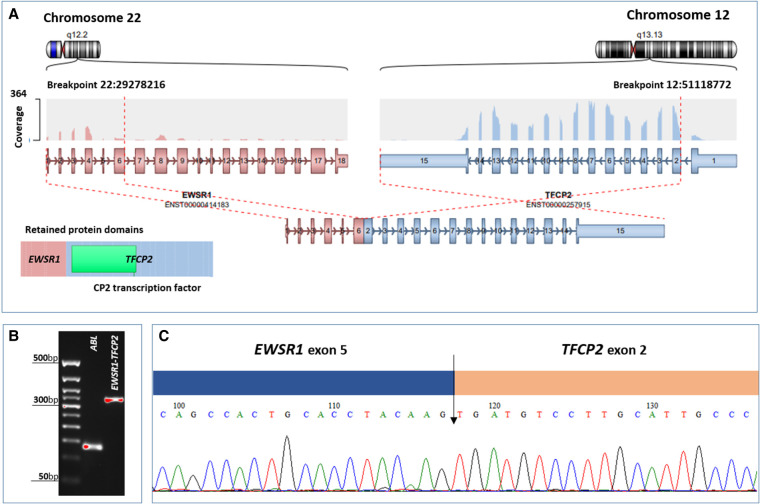
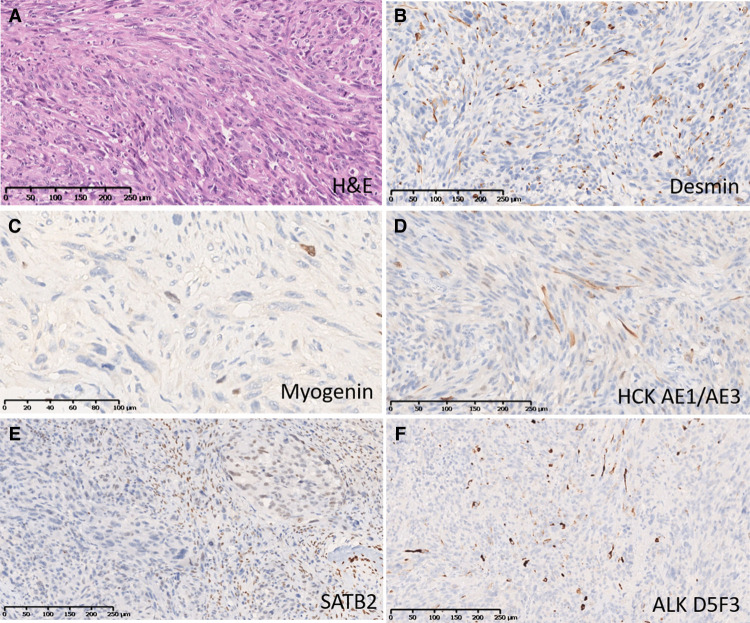
The WHO Classification of Tumors of Soft Tissue and Bone subdivides rhabdomyosarcomas (RMS) into alveolar, embryonal, pleomorphic, and spindle cell RMS. Advances in molecular genetic diagnostics have made it possible to identify new RMS subgroups within traditional morphological entities. One of these subgroups comprises rare tumors characterized by epithelioid and spindle cell morphology, highly aggressive clinical course with pronounced tendency to intraosseous growth, and the presence of pathognomonic recurring genetic aberrations- chimeric genes/transcripts EWSR1::TFCP2, FUS::TFCP2, or MEIS1::NCOA2. Starting from 2018, only 26 reported cases of RMS have been assigned to this subgroup. The rarity of such tumors hampers their correct diagnostics for both anatomic pathologists and molecular oncologists. Here we describe a clinical case of intraosseous spindle cell RMS expressing EWSR1::TFCP2 fusion gene, encountered for the first time in our practice, in a 16-year-old female patient presenting with mandibular lesion. The diagnostic process took considerable time and involved RNA sequencing; a high-throughput method of molecular genetic research. The tumor was extremely aggressive, showing resistance to polychemotherapy, radiation therapy, and crizotinib targeted therapy, with the fatal outcome.
期刊介绍:
Cold Spring Harbor Molecular Case Studies is an open-access, peer-reviewed, international journal in the field of precision medicine. Articles in the journal present genomic and molecular analyses of individuals or cohorts alongside their clinical presentations and phenotypic information. The journal''s purpose is to rapidly share insights into disease development and treatment gained by application of genomics, proteomics, metabolomics, biomarker analysis, and other approaches. The journal covers the fields of cancer, complex diseases, monogenic disorders, neurological conditions, orphan diseases, infectious disease, gene therapy, and pharmacogenomics. It has a rapid peer-review process that is based on technical evaluation of the analyses performed, not the novelty of findings, and offers a swift, clear path to publication. The journal publishes: Research Reports presenting detailed case studies of individuals and small cohorts, Research Articles describing more extensive work using larger cohorts and/or functional analyses, Rapid Communications presenting the discovery of a novel variant and/or novel phenotype associated with a known disease gene, Rapid Cancer Communications presenting the discovery of a novel variant or combination of variants in a cancer type, Variant Discrepancy Resolution describing efforts to resolve differences or update variant interpretations in ClinVar through case-level data sharing, Follow-up Reports linked to previous observations, Plus Review Articles, Editorials, and Position Statements on best practices for research in precision medicine.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
