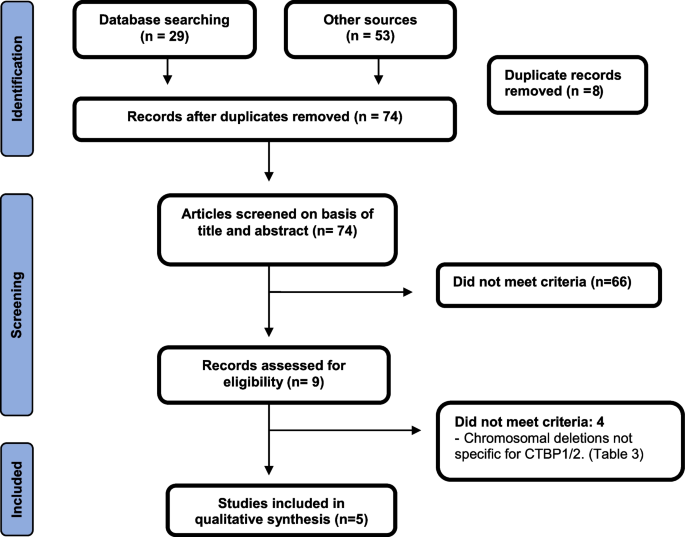
Natalia Acosta-Baena, Johanna Alexandra Tejada-Moreno, Mauricio Arcos-Burgos, Carlos Andrés Villegas-Lanau
{"title":"CTBP1 and CTBP2 mutations underpinning neurological disorders: a systematic review.","authors":"Natalia Acosta-Baena, Johanna Alexandra Tejada-Moreno, Mauricio Arcos-Burgos, Carlos Andrés Villegas-Lanau","doi":"10.1007/s10048-022-00700-w","DOIUrl":null,"url":null,"abstract":"<p><p>C-terminal binding proteins (CtBP1/2) are transcriptional coregulators that play a significant role during vertebrate neurodevelopment. This systematic review aims to identify case reports with genetic variants in CTBP1 and CTBP2 associated with brain development syndromes.We screened different databases (PubMed, Scopus, Google Scholar, LILACS) by systematically searching journals and checking reference lists and citations of background papers. We found fourteen cases (10 males) from five papers carrying two pathogenic, heterozygous variants in the CTBP1 gene (13 individuals carried the missense mutation c.991C T, p.Arg342Trp, and one subject carrying the 2-base pair deletion c.1315_1316delCA, p.Gln439ValfsTer84). These mutations were de novo in 13 cases and one case of maternal germinal mosaicism. Two variants are in the same domain of the protein: Pro-Leu-Asp-Leu-Ser (PLDLS) C terminal. Patients with these mutations exhibit a phenotype with intellectual disability, HADDTS syndrome (hypotonia, ataxia, developmental delay, and tooth enamel defects), and cerebellar volume loss. We did not identify reported cases associated with homozygous mutations harbored in CTBP1. We did not identify any report of neurodevelopment phenotypes associated with heterozygous or homozygous CTBP2 mutations. Due to CTBP2/RIBEYE being a gene with dual function, identifying and interpreting the potential pathogenic variants is challenging.Further, homozygous mutations in the CTBP2 gene may be lethal. The mechanisms involved in the pathogenesis of neurodevelopment due to variants of these proteins have not yet been elucidated, despite some functional evidence. Further studies should be conducted to understand these transcription factors and their interaction with each other and their partners.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":"23 4","pages":"231-240"},"PeriodicalIF":1.2000,"publicationDate":"2022-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9663338/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-022-00700-w","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/11/4 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
C-terminal binding proteins (CtBP1/2) are transcriptional coregulators that play a significant role during vertebrate neurodevelopment. This systematic review aims to identify case reports with genetic variants in CTBP1 and CTBP2 associated with brain development syndromes.We screened different databases (PubMed, Scopus, Google Scholar, LILACS) by systematically searching journals and checking reference lists and citations of background papers. We found fourteen cases (10 males) from five papers carrying two pathogenic, heterozygous variants in the CTBP1 gene (13 individuals carried the missense mutation c.991C T, p.Arg342Trp, and one subject carrying the 2-base pair deletion c.1315_1316delCA, p.Gln439ValfsTer84). These mutations were de novo in 13 cases and one case of maternal germinal mosaicism. Two variants are in the same domain of the protein: Pro-Leu-Asp-Leu-Ser (PLDLS) C terminal. Patients with these mutations exhibit a phenotype with intellectual disability, HADDTS syndrome (hypotonia, ataxia, developmental delay, and tooth enamel defects), and cerebellar volume loss. We did not identify reported cases associated with homozygous mutations harbored in CTBP1. We did not identify any report of neurodevelopment phenotypes associated with heterozygous or homozygous CTBP2 mutations. Due to CTBP2/RIBEYE being a gene with dual function, identifying and interpreting the potential pathogenic variants is challenging.Further, homozygous mutations in the CTBP2 gene may be lethal. The mechanisms involved in the pathogenesis of neurodevelopment due to variants of these proteins have not yet been elucidated, despite some functional evidence. Further studies should be conducted to understand these transcription factors and their interaction with each other and their partners.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
