{"title":"Insertion/deletion hotspots in the Nsp2, Nsp3, S1, and ORF8 genes of SARS-related coronaviruses.","authors":"Tetsuya Akaishi, Kei Fujiwara, Tadashi Ishii","doi":"10.1186/s12862-022-02078-7","DOIUrl":null,"url":null,"abstract":"<p><p>The genome of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) contains many insertions/deletions (indels) from the genomes of other SARS-related coronaviruses. Some of the identified indels have recently reported to involve relatively long segments of 10-300 consecutive bases and with diverse RNA sequences around gaps between virus species, both of which are different characteristics from the classical shorter in-frame indels. These non-classical complex indels have been identified in non-structural protein 3 (Nsp3), the S1 domain of the spike (S), and open reading frame 8 (ORF8). To determine whether the occurrence of these non-classical indels in specific genomic regions is ubiquitous among broad species of SARS-related coronaviruses in different animal hosts, the present study compared SARS-related coronaviruses from humans (SARS-CoV and SARS-CoV-2), bats (RaTG13 and Rc-o319), and pangolins (GX-P4L), by performing multiple sequence alignment. As a result, indel hotspots with diverse RNA sequences of different lengths between the viruses were confirmed in the Nsp2 gene (approximately 2500-2600 base positions in the overall 29,900 bases), Nsp3 gene (approximately 3000-3300 and 3800-3900 base positions), N-terminal domain of the spike protein (21,500-22,500 base positions), and ORF8 gene (27,800-28,200 base positions). Abnormally high rate of point mutations and complex indels in these regions suggest that the occurrence of mutations in these hotspots may be selectively neutral or even benefit the survival of the viruses. The presence of such indel hotspots has not been reported in different human SARS-CoV-2 strains in the last 2 years, suggesting a lower rate of indels in human SARS-CoV-2. Future studies to elucidate the mechanisms enabling the frequent development of long and complex indels in specific genomic regions of SARS-related coronaviruses would offer deeper insights into the process of viral evolution.</p>","PeriodicalId":9127,"journal":{"name":"BMC Ecology and Evolution","volume":" ","pages":"123"},"PeriodicalIF":0.0000,"publicationDate":"2022-10-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9616624/pdf/","citationCount":"6","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Ecology and Evolution","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s12862-022-02078-7","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 6
Abstract
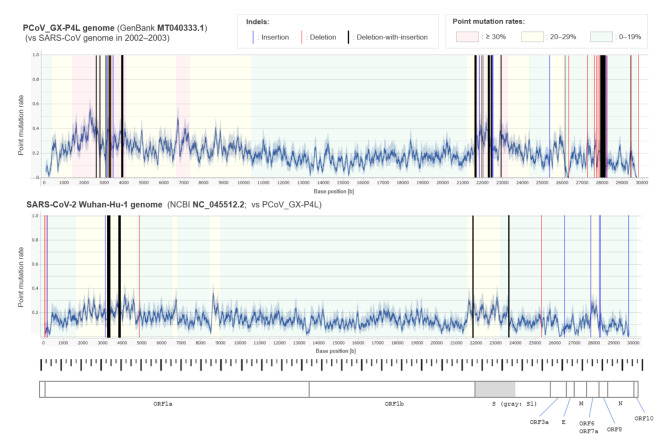
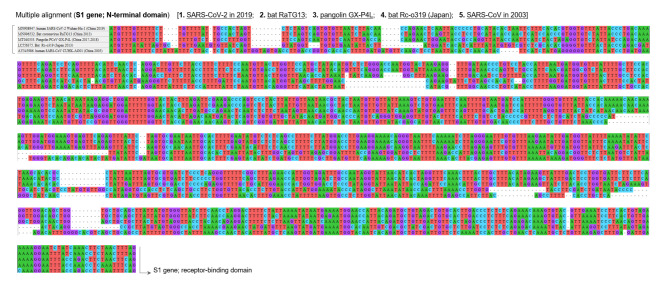
The genome of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) contains many insertions/deletions (indels) from the genomes of other SARS-related coronaviruses. Some of the identified indels have recently reported to involve relatively long segments of 10-300 consecutive bases and with diverse RNA sequences around gaps between virus species, both of which are different characteristics from the classical shorter in-frame indels. These non-classical complex indels have been identified in non-structural protein 3 (Nsp3), the S1 domain of the spike (S), and open reading frame 8 (ORF8). To determine whether the occurrence of these non-classical indels in specific genomic regions is ubiquitous among broad species of SARS-related coronaviruses in different animal hosts, the present study compared SARS-related coronaviruses from humans (SARS-CoV and SARS-CoV-2), bats (RaTG13 and Rc-o319), and pangolins (GX-P4L), by performing multiple sequence alignment. As a result, indel hotspots with diverse RNA sequences of different lengths between the viruses were confirmed in the Nsp2 gene (approximately 2500-2600 base positions in the overall 29,900 bases), Nsp3 gene (approximately 3000-3300 and 3800-3900 base positions), N-terminal domain of the spike protein (21,500-22,500 base positions), and ORF8 gene (27,800-28,200 base positions). Abnormally high rate of point mutations and complex indels in these regions suggest that the occurrence of mutations in these hotspots may be selectively neutral or even benefit the survival of the viruses. The presence of such indel hotspots has not been reported in different human SARS-CoV-2 strains in the last 2 years, suggesting a lower rate of indels in human SARS-CoV-2. Future studies to elucidate the mechanisms enabling the frequent development of long and complex indels in specific genomic regions of SARS-related coronaviruses would offer deeper insights into the process of viral evolution.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
