Prasanna Channathodiyil, Kieron May, Anne Segonds-Pichon, Paul D Smith, Simon J Cook, Jonathan Houseley
{"title":"Escape from G1 arrest during acute MEK inhibition drives the acquisition of drug resistance.","authors":"Prasanna Channathodiyil, Kieron May, Anne Segonds-Pichon, Paul D Smith, Simon J Cook, Jonathan Houseley","doi":"10.1093/narcan/zcac032","DOIUrl":null,"url":null,"abstract":"<p><p>Mutations and gene amplifications that confer drug resistance emerge frequently during chemotherapy, but their mechanism and timing are poorly understood. Here, we investigate <i>BRAF<sup>V600E</sup></i> amplification events that underlie resistance to the MEK inhibitor selumetinib (AZD6244/ARRY-142886) in COLO205 cells, a well-characterized model for reproducible emergence of drug resistance, and show that <i>BRAF</i> amplifications acquired <i>de novo</i> are the primary cause of resistance. Selumetinib causes long-term G1 arrest accompanied by reduced expression of DNA replication and repair genes, but cells stochastically re-enter the cell cycle during treatment despite continued repression of pERK1/2. Most DNA replication and repair genes are re-expressed as cells enter S and G2; however, mRNAs encoding a subset of factors important for error-free replication and chromosome segregation, including TIPIN, PLK2 and PLK3, remain at low abundance. This suggests that DNA replication following escape from G1 arrest in drug is more error prone and provides a potential explanation for the DNA damage observed under long-term RAF-MEK-ERK1/2 pathway inhibition. To test the hypothesis that escape from G1 arrest in drug promotes <i>de novo BRAF</i> amplification, we exploited the combination of palbociclib and selumetinib. Combined treatment with selumetinib and a dose of palbociclib sufficient to reinforce G1 arrest in selumetinib-sensitive cells, but not to impair proliferation of resistant cells, delays the emergence of resistant colonies, meaning that escape from G1 arrest is critical in the formation of resistant clones. Our findings demonstrate that acquisition of MEK inhibitor resistance often occurs through <i>de novo</i> gene amplification and can be suppressed by impeding cell cycle entry in drug.</p>","PeriodicalId":18879,"journal":{"name":"NAR Cancer","volume":" ","pages":"zcac032"},"PeriodicalIF":0.0000,"publicationDate":"2022-10-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9575185/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"NAR Cancer","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/narcan/zcac032","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/12/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
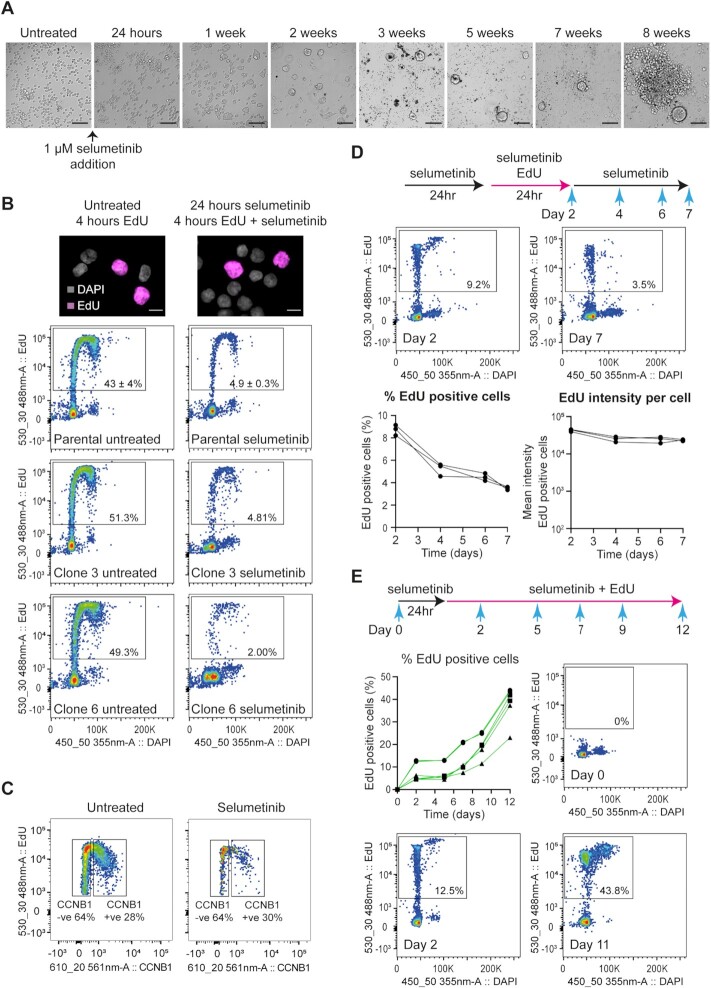
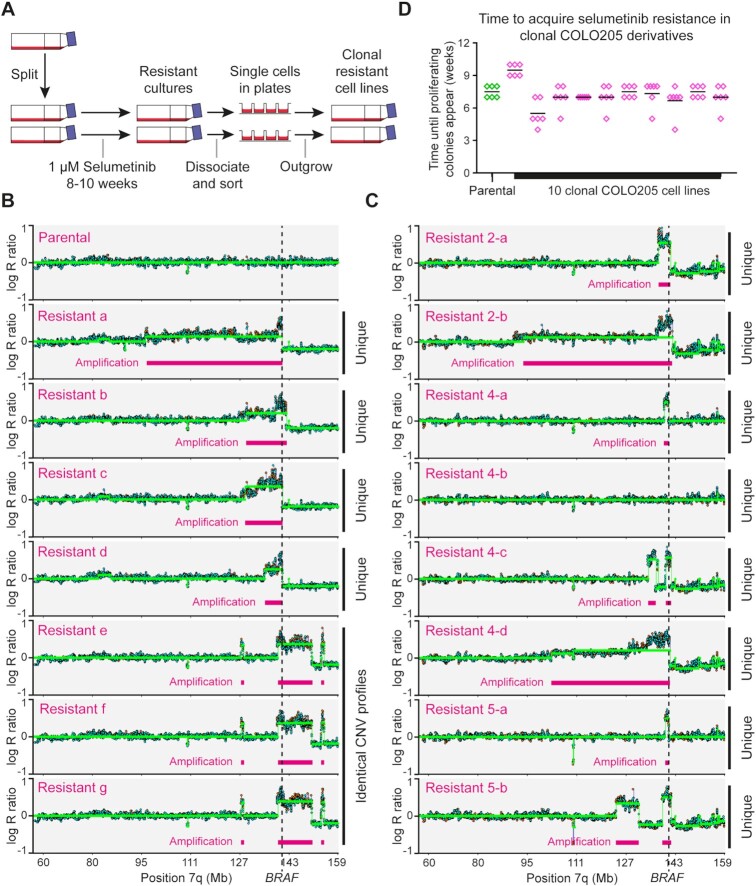
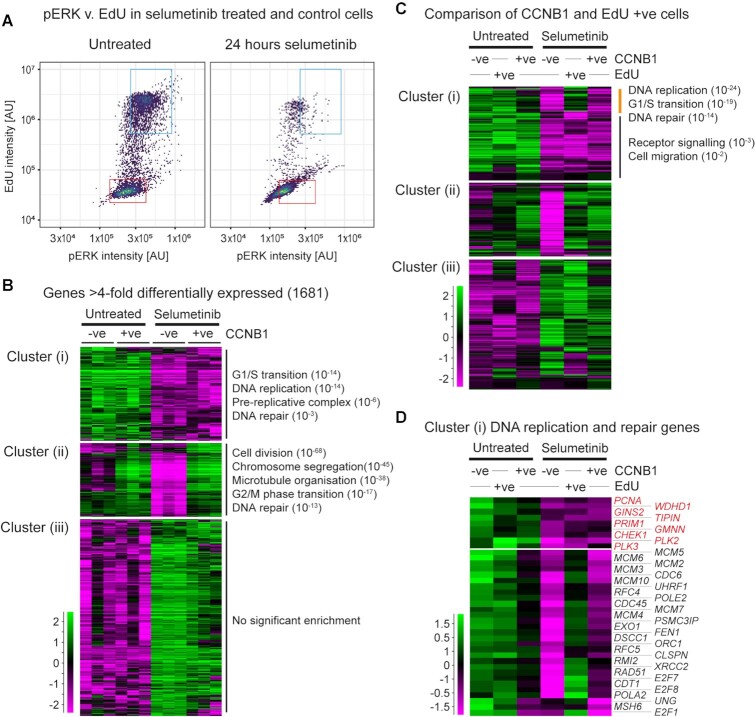
Mutations and gene amplifications that confer drug resistance emerge frequently during chemotherapy, but their mechanism and timing are poorly understood. Here, we investigate BRAFV600E amplification events that underlie resistance to the MEK inhibitor selumetinib (AZD6244/ARRY-142886) in COLO205 cells, a well-characterized model for reproducible emergence of drug resistance, and show that BRAF amplifications acquired de novo are the primary cause of resistance. Selumetinib causes long-term G1 arrest accompanied by reduced expression of DNA replication and repair genes, but cells stochastically re-enter the cell cycle during treatment despite continued repression of pERK1/2. Most DNA replication and repair genes are re-expressed as cells enter S and G2; however, mRNAs encoding a subset of factors important for error-free replication and chromosome segregation, including TIPIN, PLK2 and PLK3, remain at low abundance. This suggests that DNA replication following escape from G1 arrest in drug is more error prone and provides a potential explanation for the DNA damage observed under long-term RAF-MEK-ERK1/2 pathway inhibition. To test the hypothesis that escape from G1 arrest in drug promotes de novo BRAF amplification, we exploited the combination of palbociclib and selumetinib. Combined treatment with selumetinib and a dose of palbociclib sufficient to reinforce G1 arrest in selumetinib-sensitive cells, but not to impair proliferation of resistant cells, delays the emergence of resistant colonies, meaning that escape from G1 arrest is critical in the formation of resistant clones. Our findings demonstrate that acquisition of MEK inhibitor resistance often occurs through de novo gene amplification and can be suppressed by impeding cell cycle entry in drug.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
