{"title":"A novel mutation in GJC2 associated with hypomyelinating leukodystrophy type 2 disorder.","authors":"Sajad Rafiee Komachali, Mozhgan Sheikholeslami, Mansoor Salehi","doi":"10.5808/gi.22008","DOIUrl":null,"url":null,"abstract":"<p><p>Hypomyelinating leukodystrophy type 2 (HLD2), is an inherited genetic disease of the central nervous system caused by recessive mutations in the gap junction protein gamma 2 (GJC2/GJA12). HLD2 is characterized by nystagmus, developmental delay, motor impairments, ataxia, severe speech problem, and hypomyelination in the brain. The GJC2 sequence encodes connexin 47 protein (Cx47). Connexins are a group of membrane proteins that oligomerize to construct gap junctions protein. In the present study, a novel missense mutation gene c.760G>A (p.Val254Met) was identified in a patient with HLD2 by performing whole exome sequencing. Following the discovery of the new mutation in the proband, we used Sanger sequencing to analyze his affected sibling and parents. Sanger sequencing verified homozygosity of the mutation in the proband and his affected sibling. The autosomal recessive inheritance pattern was confirmed since Sanger sequencing revealed both healthy parents were heterozygous for the mutation. PolyPhen2, SIFT, PROVEAN, and CADD were used to evaluate the function prediction scores of detected mutations. Cx47 is essential for oligodendrocyte function, including adequate myelination and myelin maintenance in humans. Novel mutation p.Val254Met is located in the second extracellular domain of Cx47, both extracellular loops are highly conserved and probably induce intramolecular disulfide interactions. This novel mutation in the Cx47 gene causes oligodendrocyte dysfunction and HLD2 disorder.</p>","PeriodicalId":36591,"journal":{"name":"Genomics and Informatics","volume":"20 2","pages":"e24"},"PeriodicalIF":0.0000,"publicationDate":"2022-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9299563/pdf/","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genomics and Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5808/gi.22008","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/6/30 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
引用次数: 1
Abstract
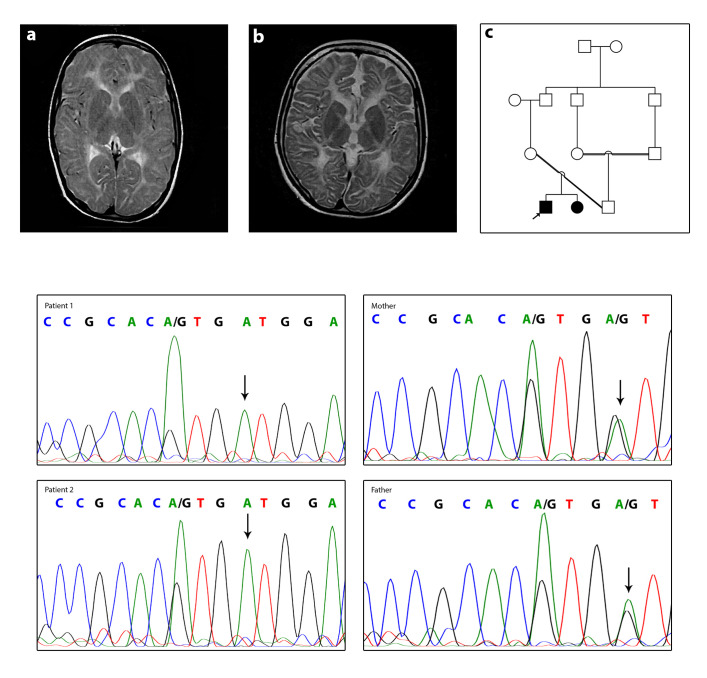
Hypomyelinating leukodystrophy type 2 (HLD2), is an inherited genetic disease of the central nervous system caused by recessive mutations in the gap junction protein gamma 2 (GJC2/GJA12). HLD2 is characterized by nystagmus, developmental delay, motor impairments, ataxia, severe speech problem, and hypomyelination in the brain. The GJC2 sequence encodes connexin 47 protein (Cx47). Connexins are a group of membrane proteins that oligomerize to construct gap junctions protein. In the present study, a novel missense mutation gene c.760G>A (p.Val254Met) was identified in a patient with HLD2 by performing whole exome sequencing. Following the discovery of the new mutation in the proband, we used Sanger sequencing to analyze his affected sibling and parents. Sanger sequencing verified homozygosity of the mutation in the proband and his affected sibling. The autosomal recessive inheritance pattern was confirmed since Sanger sequencing revealed both healthy parents were heterozygous for the mutation. PolyPhen2, SIFT, PROVEAN, and CADD were used to evaluate the function prediction scores of detected mutations. Cx47 is essential for oligodendrocyte function, including adequate myelination and myelin maintenance in humans. Novel mutation p.Val254Met is located in the second extracellular domain of Cx47, both extracellular loops are highly conserved and probably induce intramolecular disulfide interactions. This novel mutation in the Cx47 gene causes oligodendrocyte dysfunction and HLD2 disorder.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
