Ying Li, Mengyao Tang, Feng Jun Zhang, Yihan Huang, Jing Zhang, Junqi Li, Yunpeng Wang, Jinguang Yang, Shu Zhu
{"title":"Screening of ulcerative colitis biomarkers and potential pathways based on weighted gene co-expression network, machine learning and ceRNA hypothesis.","authors":"Ying Li, Mengyao Tang, Feng Jun Zhang, Yihan Huang, Jing Zhang, Junqi Li, Yunpeng Wang, Jinguang Yang, Shu Zhu","doi":"10.1186/s41065-022-00259-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Ulcerative colitis (UC) refers to an intractable intestinal inflammatory disease. Its increasing incidence rate imposes a huge burden on patients and society. The UC etiology has not been determined, so screening potential biomarkers is critical to preventing disease progression and selecting optimal therapeutic strategies more effectively.</p><p><strong>Methods: </strong>The microarray datasets of intestinal mucosal biopsy of UC patients were selected from the GEO database, and integrated with R language to screen differentially expressed genes and draw proteins interaction network diagrams. GO, KEGG, DO and GSEA enrichment analyses were performed to explore their biological functions. Through machine learning and WGCNA analysis, targets that can be used as UC potential biomarkers are screened out. ROC curves were drawn to verify the reliability of the results and predicted the mechanism of marker genes from the aspects of immune cell infiltration, co-expression analysis, and competitive endogenous network (ceRNA).</p><p><strong>Results: </strong>Two datasets GSE75214 and GSE87466 were integrated for screening, and a total of 107 differentially expressed genes were obtained. They were mainly related to biological functions such as humoral immune response and inflammatory response. Further screened out five marker genes, and found that they were associated with M0 macrophages, quiescent mast cells, M2 macrophages, and activated NK cells in terms of immune cell infiltration. The co-expression network found significant co-expression relationships between 54 miRNAs and 5 marker genes. According to the ceRNA hypothesis, NEAT1-miR-342-3p/miR-650-SLC6A14, NEAT1-miR-650-IRAK3, and XIST-miR-342-3p-IRAK3 axes were found as potential regulatory pathways in UC.</p><p><strong>Conclusion: </strong>This study screened out five biomarkers that can be used for the diagnosis and treatment of UC, namely SLC6A14, TIMP1, IRAK3, HMGCS2, and APOBEC3B. Confirmed that they play a role in the occurrence and development of UC at the level of immune infiltration, and proposed a potential RNA regulatory pathway that controls the progression of UC.</p>","PeriodicalId":12862,"journal":{"name":"Hereditas","volume":" ","pages":"42"},"PeriodicalIF":2.5000,"publicationDate":"2022-11-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9685902/pdf/","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Hereditas","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s41065-022-00259-4","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 1
Abstract
Background: Ulcerative colitis (UC) refers to an intractable intestinal inflammatory disease. Its increasing incidence rate imposes a huge burden on patients and society. The UC etiology has not been determined, so screening potential biomarkers is critical to preventing disease progression and selecting optimal therapeutic strategies more effectively.
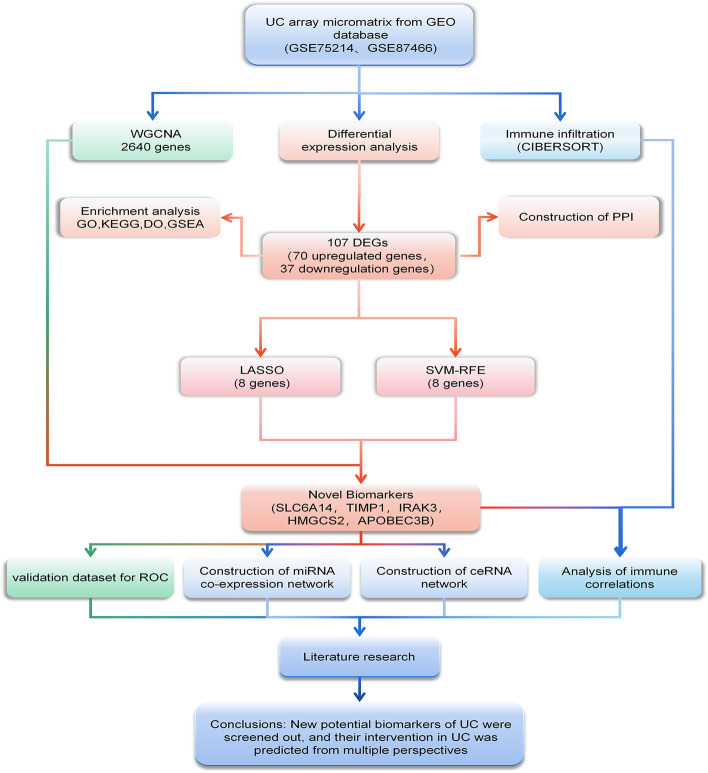
Methods: The microarray datasets of intestinal mucosal biopsy of UC patients were selected from the GEO database, and integrated with R language to screen differentially expressed genes and draw proteins interaction network diagrams. GO, KEGG, DO and GSEA enrichment analyses were performed to explore their biological functions. Through machine learning and WGCNA analysis, targets that can be used as UC potential biomarkers are screened out. ROC curves were drawn to verify the reliability of the results and predicted the mechanism of marker genes from the aspects of immune cell infiltration, co-expression analysis, and competitive endogenous network (ceRNA).
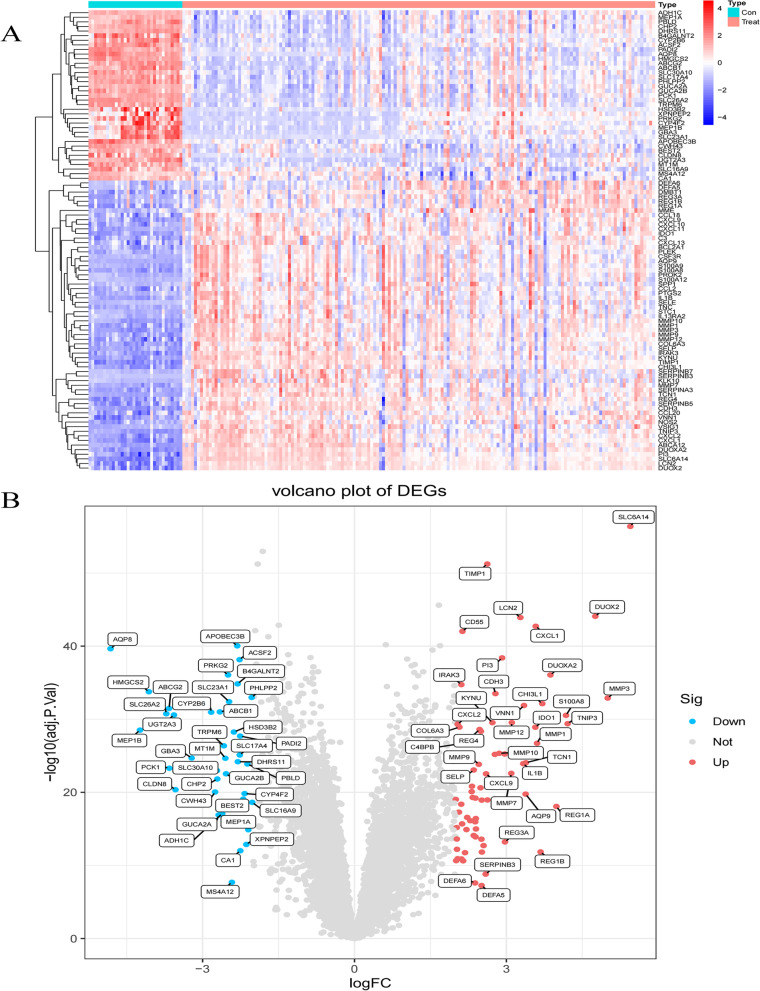
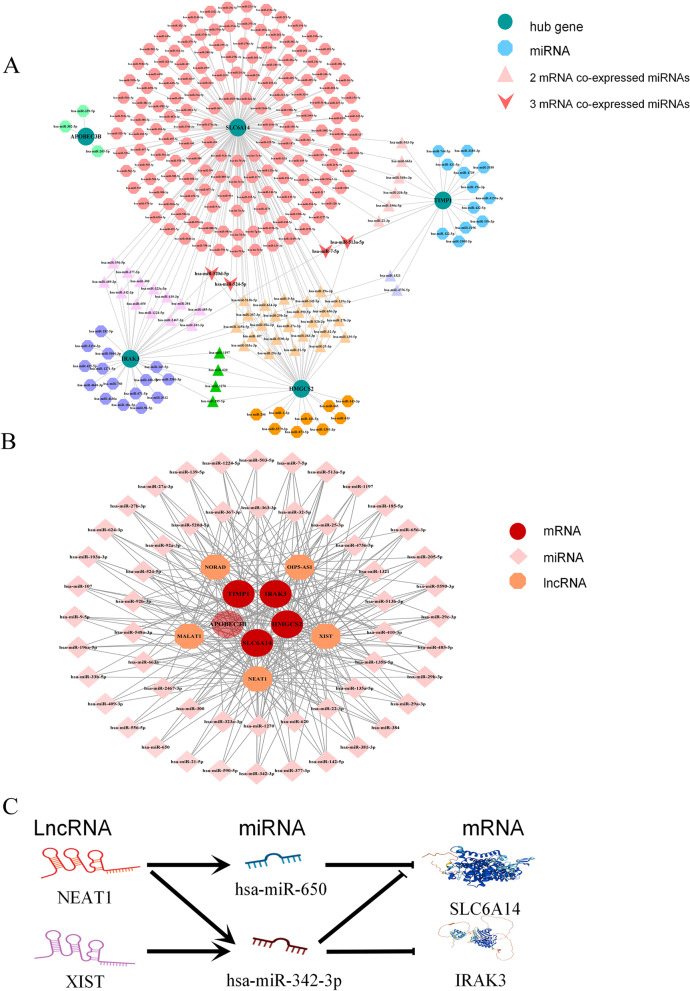
Results: Two datasets GSE75214 and GSE87466 were integrated for screening, and a total of 107 differentially expressed genes were obtained. They were mainly related to biological functions such as humoral immune response and inflammatory response. Further screened out five marker genes, and found that they were associated with M0 macrophages, quiescent mast cells, M2 macrophages, and activated NK cells in terms of immune cell infiltration. The co-expression network found significant co-expression relationships between 54 miRNAs and 5 marker genes. According to the ceRNA hypothesis, NEAT1-miR-342-3p/miR-650-SLC6A14, NEAT1-miR-650-IRAK3, and XIST-miR-342-3p-IRAK3 axes were found as potential regulatory pathways in UC.
Conclusion: This study screened out five biomarkers that can be used for the diagnosis and treatment of UC, namely SLC6A14, TIMP1, IRAK3, HMGCS2, and APOBEC3B. Confirmed that they play a role in the occurrence and development of UC at the level of immune infiltration, and proposed a potential RNA regulatory pathway that controls the progression of UC.
HereditasBiochemistry, Genetics and Molecular Biology-Genetics
CiteScore
3.80
自引率
3.70%
发文量
0
期刊介绍:
For almost a century, Hereditas has published original cutting-edge research and reviews. As the Official journal of the Mendelian Society of Lund, the journal welcomes research from across all areas of genetics and genomics. Topics of interest include human and medical genetics, animal and plant genetics, microbial genetics, agriculture and bioinformatics.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
