Dejun Zhang, Jie Wu, Yongyi Yuan, Xiaohong Li, Xue Gao, Mingyu Han, Song Gao, Shasha Huang, Pu Dai
{"title":"A novel missense variant in CEACAM16 gene causes autosomal dominant nonsyndromic hearing loss","authors":"Dejun Zhang, Jie Wu, Yongyi Yuan, Xiaohong Li, Xue Gao, Mingyu Han, Song Gao, Shasha Huang, Pu Dai","doi":"10.1111/ahg.12463","DOIUrl":null,"url":null,"abstract":"<p>Hearing loss is the most common sensorineural disorder in humans. It is estimated that more than half of cases of hearing loss are attributable to hereditary causes. Autosomal dominant nonsyndromic hearing loss (ADNSHL) is a paradigm of genetic heterogeneity with more than 45 causative genes identified to date (http://hereditaryhearingloss.org/). These genes play an essential role in the morphology and development of hair cells, synaptic transmission of the auditory nerve, and other roles in the inner ear. For example, the transmembrane channel-like gene1 (<i>TMC1</i>) is specifically required for hair cell mechanoelectrical transduction and is a functionally redundant stereocilia component (Kawashima et al., <span>2011</span>; Pan et al., <span>2013</span>) and eyes absent 4 (<i>EYA4</i>), a member of the vertebrate EYA gene family of transcriptional activators, is important for the development and maturation of the organ of Corti (Wayne et al., <span>2001</span>). However, many genes causing deafness remain unidentified (Ding et al., <span>2020</span>; Fu et al., <span>2021</span>; Qian et al., <span>2020</span>; Zhang et al., <span>2021</span>; Zhu et al., <span>2018</span>). It is therefore important to identify these genes and their functions in the inner ear.</p><p>Carcinoembryonic antigen-related cell adhesion molecule 16 (<i>CEACAM16</i>, MIM 614591) is a member of the CEACAM family, which is known to play a role in tissue architecture and homeostasis, cell growth and differentiation, angiogenesis, and tumor suppression (Kuespert et al., <span>2006</span>). <i>CEACAM16</i> maps to the DFNA4B locus on chromosome 19q13.32 and encodes a protein of 425 amino acids. Although the specific functions of the protein encoded by <i>CEACAM16</i> are still not clear, available evidence indicates that <i>CEACAM16</i> is crucial for hearing maintenance as a structural component of the tectorial membrane (Kammerer et al., <span>2012</span>). <i>CAMCAM16</i> mutations can lead to late-onset bilateral progressive sensorineural hearing loss that begin in the first or second decade (Wang et al.,2015). To date, three <i>CEACAM16</i> missense variants associated with ADNSHL have been reported (Hofrichter et al., <span>2015</span>; Wang et al., <span>2015</span>; Zheng et al., <span>2011</span>). Additionally, Booth et al. (<span>2018</span>) and Dias et al. (<span>2019</span>) described loss of function variants in <i>CEACAM16</i> that can cause autosomal recessive nonsyndromic hearing loss (ARNSHL).</p><p>In this study, we identified a novel missense variant in the <i>CEACAM16</i> gene, c.763A>G; (p.Arg255Gly) in a Chinese family using targeted next-generation sequencing approach. The phenotype was consistent with ADNSHL. Further functional experiments were carried out to evaluate the pathogenesis resulting from this mutation.</p><p>The tectorial membrane is essential for normal hearing due to its exclusive physical properties and its role in modulating the flow of fluid in the subtectorial space (Nowotny & Gummer, <span>2011</span>), amplifying the basilar membrane motion (Booth et al., <span>2019</span>; Zwislocki & Kletsky, <span>1979</span>), and shaping the cochlear tuning (Teudt & Richter, <span>2014</span>). The tectorial membrane is composed of collagen and noncollagenous proteins including alpha-tectorin (TECTA), beta-tectorin (TECTB), otogelin (OTOG), otogelin-like (OTOGL), and CEACAM16. Research in both humans and animal models have shown that pathogenic variations in genes encoding for these proteins can alter the properties of the tectorial membrane and eventually lead to hearing loss (Cheatham et al., <span>2014</span>; Ghaffari et al., <span>2013</span>; Legan et al., <span>2014</span>; Schraders et al., <span>2012</span>; Yariz et al., <span>2012</span>).</p><p>CEACAM16 protein is one of the proteins of the tectorial membrane which is thought to be essential for its function. As mentioned earlier, CEACAM16 is a secreted glycoprotein encoded by <i>CEACAM16</i> gene and belongs to the CEACAM immunoglobulin superfamily (Zheng et al., <span>2011</span>). Unlike other members of the family, CEACAM16 is well conserved in mammals and is selectively expressed in the inner ear (Kammerer et al., <span>2012</span>). The structure of the CEACAM16 protein (Figure 2b) contains two Ig constant-like domains (domain A and domain B) and two Ig variable-like domains (domain N1 and domain N2) with the N- and the C-terminus separately, and lacks both a C-terminal transmembrane domain and a glycosylphosphatidylinositol anchor (Zebhauser et al., <span>2005</span>). Its unique structure allows crosslink with TECTA and TECTB which involves the formation of the striated-sheet matrix, a laminated component that associates with the frequency-dependent mechanical properties of the tectorial membrane (Goodyear & Richardson, <span>2018</span>; Jones et al., <span>2015</span>). One study observed that the striated-sheet matrix was absent from the tectorial in the Ceacam16<sup>βgal/βgal</sup> mice (Cheatham et al., <span>2014</span>). Data from mice lacking Ceacam16 found high and low frequency hearing loss in young mice (Kammerer et al., <span>2012</span>), and in humans, a similar phenotype was reported in families harboring variants in the <i>CEACAM16</i> gene (Booth et al., <span>2018</span>; Hofrichter et al., <span>2015</span>; Wang et al., <span>2015</span>; Zheng et al., <span>2011</span>).</p><p>In our research, we identified a missense variant in <i>CEACAM16</i> (p.Arg255Gly) which caused ADNSHL in a Chinese family. All affected individuals showed bilateral progressive sensorineural hearing loss, a very similar phenotype to the one reported in an American family (1070) and another Chinese family (SY-026) (Wang et al., <span>2015</span>; Zheng et al., <span>2011</span>). However, the earliest onset age of hearing loss in the family reported in our study was 7 years old (IV-3), substantially earlier than that reported in the other families (the youngest was 10 years old) (Booth et al., <span>2018</span>; Hofrichter et al., <span>2015</span>; Wang et al., <span>2015</span>; Zheng et al., <span>2011</span>). These findings suggests that the onset of hearing loss caused by the <i>CEACAM16</i> variants can occur within the first decade of life in humans. These findings appear consistent with those seen in the Ceacam16 null mouse, which also displayed hearing loss at a young age (∼4 weeks old) (Kammerer et al., <span>2012</span>). Based on our bioinformatic analysis, the p.Arg255Gly variant cosegregated with the phenotype of DFNA4B in the family and was absent from multiple population databases (Table 1) and the 200 normal hearing controls cohort. The arginine residue at 255 in CEACAM16 was not found to be highly conserved across species (lack of conservation in dogs) (Figure 2A). The p.Arg255Gly variant is located within the Ig constant-like domain B (Figure 2B), which is crucial for stabilizing the Ig-like conformation and increasing the affinity of the homophilic binding (Athanasia & Andreas, <span>2014</span>; Bonsor et al., <span>2015</span>). Therefore, we speculate that this novel missense variant might damage the structure and function of CEACAM16.</p><p>Transfection of the mutant protein (p.Arg255Gly) in HEK293T cells, visualized using immunofluorescence, suggest similar intracellular and extracellular protein expression between mutant and wild type proteins. These results are in agreement with our findings using Western blot, and suggest that the p.Arg255Gly variant does not interfere with the normal subcellular localization of CEACAM16. Our findings are also consistent with those reported by Wang et al. (<span>2015</span>) and Zheng et al. (<span>2011</span>). Furthermore, we performed quantitative analysis of the mutant proteins extracted from the cell lysate and culture medium by Western blot and ELISA, respectively. Interestingly, in both cases the amount of mutant protein was higher than that of WT, indicating that the new variant in <i>CEACAM16</i> increases the secretion of the mutant CEACAM16 protein, and suggesting it is a gain-of-function mutation. Subsequently, in order to investigate whether the variant p.Arg255Gly affected protein expression at the transcription level, we used PCR to detect the relative expression l <i>CEACAM16</i> mRNA level in HEK293T cells. In contrast to the Western blot results, PCR results indicated that mRNA expression levels of mutant <i>CEACAM16</i> were significantly lower than those of the WT group. There may be several reasons for this inconsistency. First, the difference in plasmid transfection efficiency of transiently transfected HEK293T cells may have masked the true mRNA levels of the mutant gene. Second, according to the research of Perl et al. (<span>2017</span>), protein levels are more conserved than mRNA levels in all datasets, and changes in transcription are associated with translational changes that exert opposite effects on the protein level. Third, some other factors may influence the correlation between mRNA and protein expression, such as mRNA degradation rate, variation of mRNA secondary structure, among others (Guo et al., <span>2008</span>). To date, the specific function of the protein encoded by <i>CEACAM16</i>, as well as the pathogenetic mechanisms underlying hearing impairment of patients harboring variants in this gene, remain unclear. In previous studies, a missense variant (p.Thr140Pro) in <i>CEACAM16</i> was identified in an ADNSHL pedigree and was predicted to disrupt a glycosylation site, interfering with protein stability (Zheng et al., <span>2011</span>). In another family with hearing loss, the p.Gly169Arg variant at Ig-C like domain A was assumed to change the spatial structure of CEACAM16 and decrease stability and the polymerizing ability of the protein (Wang et al., <span>2015</span>). However, these studies lack in vivo evidence to help validate their assumptions. To the best of our knowledge, CEACAM16 is a secreted glycoprotein with a signal peptide at the N terminus; guided by the signal peptide, the newly synthesized glycoprotein is translocated to the endoplasmic reticulum and then secreted (Blobel & Dobberstein, <span>1975</span>). According to Kammerer et al. (<span>2012</span>), CEACAM16 is located in Deiters and interdental cells of the organ of Corti in the inner ear of mice, and released via the Deiters cell's projections, after which is incorporated into the matrix of the tectorial membrane. In the tectorial membrane, CEACAM16 interacts with TECTA and TECTB via its two Ig-V like domains where it forms the dark and light zone of the striated-sheet matrix (Goodyear & Richardson, <span>2018</span>). The p.Arg255Gly mutation we report occurs within the Ig-C like domain B of CEACAM16. This Ig-C like domain contains conserved cysteine residues that stabilize the Ig-like conformation by forming a disulphide bridge (Bork et al., <span>1994</span>; Williams & Barclay, <span>1988</span>), and plays a key role in CEACAM16 dimer formation (Kammerer et al., <span>2012</span>). By comparing the properties of arginine and glycine, we know that the mutant residue is smaller and more hydrophobic than the wild-type residue, and is neutral in contrast to the wild-type residue, which is negatively charged. This mutation introduces an amino acid with distinct properties, which can disturb the Ig-C like domain B and decrease protein stability (Cheng et al., <span>2006</span>; Venselaar et al., <span>2010</span>). Therefore, we speculate that the variant p.Arg255Gly might cause structural changes in the CEACAM16 protein, altering its intracellular synthesis and transport pathways, and leading to increased secretion. At the same time, due to the decreased stability of the extracellular protein, the mutation might interfere with CEACAM16 dimerization and affect the interaction between CEACAM16 and other proteins. This alteration might also compromise the physical properties of the tectorial membrane and eventually lead to hearing loss. This study also has some limitations. We used CEACAM16 antibody commercial kits and were unable to detect oligomers using Western blot. Further vivo experiments are necessary to investigate the pathophysiological significance of CEACAM16.</p><p>In summary, we implicate a novel variant in <i>CEACAM16</i> as the genetic cause of progressive hearing loss in a large Chinese pedigree. We suggest that this variant leads to increased secretion of mutant CEACAM16 protein in vitro, uncovering a putative novel mechanism underlying DFNA4B-related hearing loss.</p><p>The authors declare no conflict of interest.</p><p>Dejun Zhang, Shasha Huang, and Pu Dai carried out the studies, participated in collecting data, and drafted the manuscript. Jie Wu, Yongyi Yuan, and Xiaohong Li performed the statistical analysis and participated in its design. Xue Gao, Mingyu Han, and Song Gao helped to draft the manuscript and performed the data analysis. All authors read and approved the final manuscript.</p>","PeriodicalId":8085,"journal":{"name":"Annals of Human Genetics","volume":"86 4","pages":"207-217"},"PeriodicalIF":1.2000,"publicationDate":"2022-03-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9314904/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Annals of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/ahg.12463","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Hearing loss is the most common sensorineural disorder in humans. It is estimated that more than half of cases of hearing loss are attributable to hereditary causes. Autosomal dominant nonsyndromic hearing loss (ADNSHL) is a paradigm of genetic heterogeneity with more than 45 causative genes identified to date (http://hereditaryhearingloss.org/). These genes play an essential role in the morphology and development of hair cells, synaptic transmission of the auditory nerve, and other roles in the inner ear. For example, the transmembrane channel-like gene1 (TMC1) is specifically required for hair cell mechanoelectrical transduction and is a functionally redundant stereocilia component (Kawashima et al., 2011; Pan et al., 2013) and eyes absent 4 (EYA4), a member of the vertebrate EYA gene family of transcriptional activators, is important for the development and maturation of the organ of Corti (Wayne et al., 2001). However, many genes causing deafness remain unidentified (Ding et al., 2020; Fu et al., 2021; Qian et al., 2020; Zhang et al., 2021; Zhu et al., 2018). It is therefore important to identify these genes and their functions in the inner ear.
Carcinoembryonic antigen-related cell adhesion molecule 16 (CEACAM16, MIM 614591) is a member of the CEACAM family, which is known to play a role in tissue architecture and homeostasis, cell growth and differentiation, angiogenesis, and tumor suppression (Kuespert et al., 2006). CEACAM16 maps to the DFNA4B locus on chromosome 19q13.32 and encodes a protein of 425 amino acids. Although the specific functions of the protein encoded by CEACAM16 are still not clear, available evidence indicates that CEACAM16 is crucial for hearing maintenance as a structural component of the tectorial membrane (Kammerer et al., 2012). CAMCAM16 mutations can lead to late-onset bilateral progressive sensorineural hearing loss that begin in the first or second decade (Wang et al.,2015). To date, three CEACAM16 missense variants associated with ADNSHL have been reported (Hofrichter et al., 2015; Wang et al., 2015; Zheng et al., 2011). Additionally, Booth et al. (2018) and Dias et al. (2019) described loss of function variants in CEACAM16 that can cause autosomal recessive nonsyndromic hearing loss (ARNSHL).
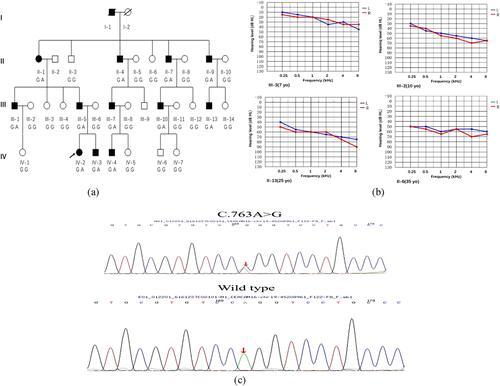
In this study, we identified a novel missense variant in the CEACAM16 gene, c.763A>G; (p.Arg255Gly) in a Chinese family using targeted next-generation sequencing approach. The phenotype was consistent with ADNSHL. Further functional experiments were carried out to evaluate the pathogenesis resulting from this mutation.
The tectorial membrane is essential for normal hearing due to its exclusive physical properties and its role in modulating the flow of fluid in the subtectorial space (Nowotny & Gummer, 2011), amplifying the basilar membrane motion (Booth et al., 2019; Zwislocki & Kletsky, 1979), and shaping the cochlear tuning (Teudt & Richter, 2014). The tectorial membrane is composed of collagen and noncollagenous proteins including alpha-tectorin (TECTA), beta-tectorin (TECTB), otogelin (OTOG), otogelin-like (OTOGL), and CEACAM16. Research in both humans and animal models have shown that pathogenic variations in genes encoding for these proteins can alter the properties of the tectorial membrane and eventually lead to hearing loss (Cheatham et al., 2014; Ghaffari et al., 2013; Legan et al., 2014; Schraders et al., 2012; Yariz et al., 2012).
CEACAM16 protein is one of the proteins of the tectorial membrane which is thought to be essential for its function. As mentioned earlier, CEACAM16 is a secreted glycoprotein encoded by CEACAM16 gene and belongs to the CEACAM immunoglobulin superfamily (Zheng et al., 2011). Unlike other members of the family, CEACAM16 is well conserved in mammals and is selectively expressed in the inner ear (Kammerer et al., 2012). The structure of the CEACAM16 protein (Figure 2b) contains two Ig constant-like domains (domain A and domain B) and two Ig variable-like domains (domain N1 and domain N2) with the N- and the C-terminus separately, and lacks both a C-terminal transmembrane domain and a glycosylphosphatidylinositol anchor (Zebhauser et al., 2005). Its unique structure allows crosslink with TECTA and TECTB which involves the formation of the striated-sheet matrix, a laminated component that associates with the frequency-dependent mechanical properties of the tectorial membrane (Goodyear & Richardson, 2018; Jones et al., 2015). One study observed that the striated-sheet matrix was absent from the tectorial in the Ceacam16βgal/βgal mice (Cheatham et al., 2014). Data from mice lacking Ceacam16 found high and low frequency hearing loss in young mice (Kammerer et al., 2012), and in humans, a similar phenotype was reported in families harboring variants in the CEACAM16 gene (Booth et al., 2018; Hofrichter et al., 2015; Wang et al., 2015; Zheng et al., 2011).
In our research, we identified a missense variant in CEACAM16 (p.Arg255Gly) which caused ADNSHL in a Chinese family. All affected individuals showed bilateral progressive sensorineural hearing loss, a very similar phenotype to the one reported in an American family (1070) and another Chinese family (SY-026) (Wang et al., 2015; Zheng et al., 2011). However, the earliest onset age of hearing loss in the family reported in our study was 7 years old (IV-3), substantially earlier than that reported in the other families (the youngest was 10 years old) (Booth et al., 2018; Hofrichter et al., 2015; Wang et al., 2015; Zheng et al., 2011). These findings suggests that the onset of hearing loss caused by the CEACAM16 variants can occur within the first decade of life in humans. These findings appear consistent with those seen in the Ceacam16 null mouse, which also displayed hearing loss at a young age (∼4 weeks old) (Kammerer et al., 2012). Based on our bioinformatic analysis, the p.Arg255Gly variant cosegregated with the phenotype of DFNA4B in the family and was absent from multiple population databases (Table 1) and the 200 normal hearing controls cohort. The arginine residue at 255 in CEACAM16 was not found to be highly conserved across species (lack of conservation in dogs) (Figure 2A). The p.Arg255Gly variant is located within the Ig constant-like domain B (Figure 2B), which is crucial for stabilizing the Ig-like conformation and increasing the affinity of the homophilic binding (Athanasia & Andreas, 2014; Bonsor et al., 2015). Therefore, we speculate that this novel missense variant might damage the structure and function of CEACAM16.
Transfection of the mutant protein (p.Arg255Gly) in HEK293T cells, visualized using immunofluorescence, suggest similar intracellular and extracellular protein expression between mutant and wild type proteins. These results are in agreement with our findings using Western blot, and suggest that the p.Arg255Gly variant does not interfere with the normal subcellular localization of CEACAM16. Our findings are also consistent with those reported by Wang et al. (2015) and Zheng et al. (2011). Furthermore, we performed quantitative analysis of the mutant proteins extracted from the cell lysate and culture medium by Western blot and ELISA, respectively. Interestingly, in both cases the amount of mutant protein was higher than that of WT, indicating that the new variant in CEACAM16 increases the secretion of the mutant CEACAM16 protein, and suggesting it is a gain-of-function mutation. Subsequently, in order to investigate whether the variant p.Arg255Gly affected protein expression at the transcription level, we used PCR to detect the relative expression l CEACAM16 mRNA level in HEK293T cells. In contrast to the Western blot results, PCR results indicated that mRNA expression levels of mutant CEACAM16 were significantly lower than those of the WT group. There may be several reasons for this inconsistency. First, the difference in plasmid transfection efficiency of transiently transfected HEK293T cells may have masked the true mRNA levels of the mutant gene. Second, according to the research of Perl et al. (2017), protein levels are more conserved than mRNA levels in all datasets, and changes in transcription are associated with translational changes that exert opposite effects on the protein level. Third, some other factors may influence the correlation between mRNA and protein expression, such as mRNA degradation rate, variation of mRNA secondary structure, among others (Guo et al., 2008). To date, the specific function of the protein encoded by CEACAM16, as well as the pathogenetic mechanisms underlying hearing impairment of patients harboring variants in this gene, remain unclear. In previous studies, a missense variant (p.Thr140Pro) in CEACAM16 was identified in an ADNSHL pedigree and was predicted to disrupt a glycosylation site, interfering with protein stability (Zheng et al., 2011). In another family with hearing loss, the p.Gly169Arg variant at Ig-C like domain A was assumed to change the spatial structure of CEACAM16 and decrease stability and the polymerizing ability of the protein (Wang et al., 2015). However, these studies lack in vivo evidence to help validate their assumptions. To the best of our knowledge, CEACAM16 is a secreted glycoprotein with a signal peptide at the N terminus; guided by the signal peptide, the newly synthesized glycoprotein is translocated to the endoplasmic reticulum and then secreted (Blobel & Dobberstein, 1975). According to Kammerer et al. (2012), CEACAM16 is located in Deiters and interdental cells of the organ of Corti in the inner ear of mice, and released via the Deiters cell's projections, after which is incorporated into the matrix of the tectorial membrane. In the tectorial membrane, CEACAM16 interacts with TECTA and TECTB via its two Ig-V like domains where it forms the dark and light zone of the striated-sheet matrix (Goodyear & Richardson, 2018). The p.Arg255Gly mutation we report occurs within the Ig-C like domain B of CEACAM16. This Ig-C like domain contains conserved cysteine residues that stabilize the Ig-like conformation by forming a disulphide bridge (Bork et al., 1994; Williams & Barclay, 1988), and plays a key role in CEACAM16 dimer formation (Kammerer et al., 2012). By comparing the properties of arginine and glycine, we know that the mutant residue is smaller and more hydrophobic than the wild-type residue, and is neutral in contrast to the wild-type residue, which is negatively charged. This mutation introduces an amino acid with distinct properties, which can disturb the Ig-C like domain B and decrease protein stability (Cheng et al., 2006; Venselaar et al., 2010). Therefore, we speculate that the variant p.Arg255Gly might cause structural changes in the CEACAM16 protein, altering its intracellular synthesis and transport pathways, and leading to increased secretion. At the same time, due to the decreased stability of the extracellular protein, the mutation might interfere with CEACAM16 dimerization and affect the interaction between CEACAM16 and other proteins. This alteration might also compromise the physical properties of the tectorial membrane and eventually lead to hearing loss. This study also has some limitations. We used CEACAM16 antibody commercial kits and were unable to detect oligomers using Western blot. Further vivo experiments are necessary to investigate the pathophysiological significance of CEACAM16.
In summary, we implicate a novel variant in CEACAM16 as the genetic cause of progressive hearing loss in a large Chinese pedigree. We suggest that this variant leads to increased secretion of mutant CEACAM16 protein in vitro, uncovering a putative novel mechanism underlying DFNA4B-related hearing loss.
The authors declare no conflict of interest.
Dejun Zhang, Shasha Huang, and Pu Dai carried out the studies, participated in collecting data, and drafted the manuscript. Jie Wu, Yongyi Yuan, and Xiaohong Li performed the statistical analysis and participated in its design. Xue Gao, Mingyu Han, and Song Gao helped to draft the manuscript and performed the data analysis. All authors read and approved the final manuscript.
听力损失是人类最常见的感觉神经障碍。据估计,半数以上的听力损失病例可归因于遗传原因。常染色体显性非综合征性听力损失(ADNSHL)是一种遗传异质性的范例,迄今已确定的致病基因超过45个(http://hereditaryhearingloss.org/)。这些基因在毛细胞的形态和发育、听神经的突触传递以及内耳的其他功能中发挥着重要作用。例如,跨膜通道样基因1 (TMC1)是毛细胞机电转导所必需的,是一个功能冗余的立体纤毛成分(Kawashima et al., 2011;Pan et al., 2013)和eyes absent 4 (EYA4),是脊椎动物EYA基因转录激活因子家族的一员,对Corti器官的发育和成熟很重要(Wayne et al., 2001)。然而,许多导致耳聋的基因仍未被识别(Ding et al., 2020;Fu et al., 2021;Qian et al., 2020;Zhang et al., 2021;Zhu et al., 2018)。因此,确定这些基因及其在内耳中的功能是很重要的。癌胚抗原相关细胞粘附分子16 (CEACAM16, MIM 614591)是CEACAM家族的一员,已知在组织构建和稳态、细胞生长和分化、血管生成和肿瘤抑制中发挥作用(Kuespert et al., 2006)。CEACAM16定位于染色体19q13.32上的DFNA4B位点,编码425个氨基酸的蛋白质。虽然CEACAM16编码蛋白的具体功能尚不清楚,但现有证据表明,CEACAM16作为被膜的结构成分,对听力维持至关重要(Kammerer et al., 2012)。CAMCAM16突变可导致晚发性双侧进行性感音神经性听力损失,始于第一或第二个十年(Wang et al.,2015)。迄今为止,已经报道了三种与ADNSHL相关的CEACAM16错义变体(Hofrichter et al., 2015;Wang et al., 2015;郑等人,2011)。此外,Booth等人(2018)和Dias等人(2019)描述了CEACAM16中可导致常染色体隐性非综合征性听力损失(ARNSHL)的功能变异丧失。在这项研究中,我们在CEACAM16基因中发现了一个新的错义变体c.763A>G;(p.a g255gly)在中国家庭中使用靶向下一代测序方法。表型与ADNSHL一致。我们进行了进一步的功能实验来评估这种突变的发病机制。由于其独特的物理特性及其在调节膜下空间液体流动方面的作用,被膜对正常听力是必不可少的(Nowotny &Gummer, 2011),放大基底膜运动(Booth et al., 2019;Zwislocki,Kletsky, 1979)和塑造耳蜗调谐(Teudt &里希特,2014)。被膜由胶原蛋白和非胶原蛋白组成,包括α - TECTA (TECTA)、β - TECTB (TECTB)、otogelin (OTOG)、otogelin-like (OTOGL)和CEACAM16。人类和动物模型的研究表明,编码这些蛋白质的基因的致病变异可以改变被膜的特性,最终导致听力损失(Cheatham et al., 2014;Ghaffari et al., 2013;Legan et al., 2014;Schraders et al., 2012;Yariz et al., 2012)。CEACAM16蛋白是被膜上的一种蛋白质,被认为对其功能至关重要。如前所述,CEACAM16是由CEACAM16基因编码的分泌糖蛋白,属于CEACAM免疫球蛋白超家族(Zheng et al., 2011)。与该家族的其他成员不同,CEACAM16在哺乳动物中保守性较好,并在内耳中选择性表达(Kammerer et al., 2012)。CEACAM16蛋白的结构(图2b)包含两个Ig恒定结构域(结构域A和结构域B)和两个Ig可变结构域(结构域N1和结构域N2),分别位于N端和c端,缺乏c端跨膜结构域和糖基磷脂酰肌醇锚定结构域(Zebhauser et al., 2005)。其独特的结构允许与TECTA和TECTB进行交联,这涉及到条纹片基质的形成,这是一种层压组件,与膜的频率相关的机械性能(Goodyear &理查森,2018;Jones et al., 2015)。一项研究发现,Ceacam16βgal/βgal小鼠的皮层中不存在条纹片基质(Cheatham et al., 2014)。来自缺乏Ceacam16的小鼠的数据发现,年轻小鼠的高频和低频听力损失(Kammerer等人,2012),在人类中,在Ceacam16基因变异的家族中报告了类似的表型(Booth等人,2018;Hofrichter et al., 2015;Wang et al., 2015;郑等人,2011)。在我们的研究中,我们在CEACAM16中发现了一个错义变体(p。 Arg255Gly)在一个中国家庭中引起ADNSHL。所有受影响的个体均表现为双侧进行性感音神经性听力损失,这一表型与一个美国家庭(1070)和另一个中国家庭(y -026)的报告非常相似(Wang et al., 2015;郑等人,2011)。然而,我们研究中报告的家庭中听力损失的最早发病年龄为7岁(IV-3岁),大大早于其他家庭报告的年龄(最小的为10岁)(Booth et al., 2018;Hofrichter et al., 2015;Wang et al., 2015;郑等人,2011)。这些发现表明,由CEACAM16变异引起的听力损失可能发生在人类生命的头十年。这些发现似乎与在Ceacam16缺失小鼠中观察到的结果一致,这些小鼠在幼年(~ 4周龄)也表现出听力损失(Kammerer et al., 2012)。根据我们的生物信息学分析,p.a g255gly变异与家族中DFNA4B表型共分离,并且在多个人群数据库(表1)和200名正常听力对照队列中缺失。CEACAM16中255处的精氨酸残基在物种间没有高度保守性(狗中缺乏保守性)(图2A)。p.Arg255Gly变体位于Ig恒定结构域B内(图2B),这对于稳定Ig样构象和增加亲同源结合的亲和力至关重要(Athanasia &安德烈亚斯,2014;Bonsor et al., 2015)。因此,我们推测这种新的错义变体可能会破坏CEACAM16的结构和功能。在HEK293T细胞中转染突变蛋白(p.a g255gly),使用免疫荧光显示突变型蛋白和野生型蛋白在细胞内和细胞外表达相似。这些结果与我们使用Western blot的发现一致,表明p.a g255gly变异不会干扰CEACAM16正常的亚细胞定位。我们的研究结果也与Wang et al.(2015)和Zheng et al.(2011)的报告一致。此外,我们分别用Western blot和ELISA对从细胞裂解液和培养基中提取的突变蛋白进行了定量分析。有趣的是,在这两种情况下,突变蛋白的数量都高于WT,这表明CEACAM16的新变体增加了突变CEACAM16蛋白的分泌,这表明它是一种功能获得突变。随后,为了研究p.a g255gly变异是否在转录水平上影响蛋白表达,我们采用PCR检测HEK293T细胞中CEACAM16 mRNA的相对表达水平。与Western blot结果相比,PCR结果显示突变体CEACAM16 mRNA表达水平显著低于WT组。这种不一致可能有几个原因。首先,瞬时转染的HEK293T细胞质粒转染效率的差异可能掩盖了突变基因的真实mRNA水平。其次,根据Perl等人(2017)的研究,在所有数据集中,蛋白质水平比mRNA水平更保守,转录的变化与翻译变化相关,而翻译变化对蛋白质水平产生相反的影响。第三,其他一些因素可能会影响mRNA与蛋白质表达的相关性,如mRNA降解率、mRNA二级结构的变化等(Guo et al., 2008)。迄今为止,CEACAM16编码蛋白的具体功能以及携带该基因变异的患者听力障碍的发病机制尚不清楚。在之前的研究中,在一个ADNSHL家系中发现了CEACAM16的一个错义变体(p.s thr140pro),预计它会破坏一个糖基化位点,干扰蛋白质的稳定性(Zheng et al., 2011)。在另一个听力损失家族中,p.Gly169Arg在Ig-C样结构域A的变异被认为改变了CEACAM16的空间结构,降低了该蛋白的稳定性和聚合能力(Wang et al., 2015)。然而,这些研究缺乏体内证据来帮助验证他们的假设。据我们所知,CEACAM16是一种分泌糖蛋白,在N端有一个信号肽;在信号肽的引导下,新合成的糖蛋白转运到内质网并分泌(Blobel &Dobberstein, 1975)。据Kammerer et al.(2012)研究,CEACAM16位于小鼠内耳Corti器官的deiter细胞和牙间细胞中,通过deiter细胞的突起释放,然后被纳入到被膜基质中。在覆盖膜中,CEACAM16通过其两个Ig-V样结构域与TECTA和TECTB相互作用,形成条纹片基质的暗区和亮区(Goodyear &理查森,2018)。p。 我们报道的Arg255Gly突变发生在CEACAM16的Ig-C样结构域B内。这种类igg - c结构域含有保守的半胱氨酸残基,通过形成二硫桥来稳定类igg构象(Bork等,1994;威廉姆斯,Barclay, 1988),并在CEACAM16二聚体形成中起关键作用(Kammerer et al., 2012)。通过比较精氨酸和甘氨酸的性质,我们知道突变残基比野生型残基更小,更疏水,与带负电的野生型残基相比,突变残基是中性的。这种突变引入了一种具有不同性质的氨基酸,它可以扰乱Ig-C样结构域B并降低蛋白质稳定性(Cheng et al., 2006;Venselaar et al., 2010)。因此,我们推测变异p.a g255gly可能引起CEACAM16蛋白的结构改变,改变其胞内合成和转运途径,导致分泌增加。同时,由于胞外蛋白稳定性下降,突变可能干扰CEACAM16二聚化,影响CEACAM16与其他蛋白的相互作用。这种改变也可能损害被膜的物理特性,最终导致听力丧失。本研究也有一定的局限性。我们使用CEACAM16抗体商用试剂盒,无法使用Western blot检测低聚物。CEACAM16的病理生理意义有待进一步的体内实验研究。综上所述,我们认为CEACAM16基因的一种新变异可能是导致中国大型家系进行性听力损失的遗传原因。我们认为这种变异导致体外CEACAM16突变蛋白分泌增加,揭示了dfna4b相关听力损失的新机制。作者声明无利益冲突。张德军、黄莎莎、戴璞负责研究,参与数据收集,撰写论文。吴杰、袁永义、李晓红进行统计分析并参与设计。高雪、韩明宇、高松协助撰写稿件并进行数据分析。所有作者都阅读并批准了最终的手稿。
期刊介绍:
Annals of Human Genetics publishes material directly concerned with human genetics or the application of scientific principles and techniques to any aspect of human inheritance. Papers that describe work on other species that may be relevant to human genetics will also be considered. Mathematical models should include examples of application to data where possible.
Authors are welcome to submit Supporting Information, such as data sets or additional figures or tables, that will not be published in the print edition of the journal, but which will be viewable via the online edition and stored on the website.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
