Mutational screening of Greek patients with axonal Charcot-Marie-Tooth disease using targeted next-generation sequencing: Clinical and molecular spectrum delineation
{"title":"Mutational screening of Greek patients with axonal Charcot-Marie-Tooth disease using targeted next-generation sequencing: Clinical and molecular spectrum delineation","authors":"Zoi Kontogeorgiou, Chrisoula Kartanou, Michail Rentzos, Panagiotis Kokotis, Evangelos Anagnostou, Thomas Zambelis, Elisabeth Chroni, Argyris Dinopoulos, Marios Panas, Georgios Koutsis, Georgia Karadima","doi":"10.1111/jns.12598","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Background and Aims</h3>\n \n <p>Axonal forms of Charcot-Marie-Tooth disease (CMT) are classified as CMT2, distal hereditary motor neuropathy (dHMN) or hereditary sensory neuropathy (HSN) and can be caused by mutations in over 100 genes. We presently aimed to investigate for the first time the genetic landscape of axonal CMT in the Greek population.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>Sixty index patients with CMT2, dHMN or HSN were screened by a combination of Sanger sequencing (<i>GJB1</i>) and next-generation sequencing custom-made gene panel covering 24 commonly mutated genes in axonal CMT.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>Overall, 20 variants classified as pathogenic or likely pathogenic were identified in heterozygous state in 20 index cases, representing 33.3% of the cohort. Of these, 14 were known pathogenic/likely pathogenic and six were designated as such according to ACMG classification, after in silico evaluation, testing for familial segregation and further literature review. The most frequently involved genes were <i>GJB1</i> (11.7%), <i>MPZ</i> (5%) and <i>MFN2</i> (5%), followed by <i>DNM2</i> (3.3%) and <i>LRSAM1</i> (3.3%). Single cases were identified with mutations in <i>BSCL2</i>, <i>HSPB1</i> and <i>GDAP1</i>.</p>\n </section>\n \n <section>\n \n <h3> Interpretation</h3>\n \n <p>A wide phenotypic variability in terms of severity and age of onset was noted. Given the limited number of genes tested, the diagnostic yield of the present panel compares favourably with studies in other European populations. Our study delineates the genetic and phenotypic variability of inherited axonal neuropathies in the Greek population and contributes to the pathogenicity characterization of further variants linked to axonal neuropathies.</p>\n </section>\n </div>","PeriodicalId":17451,"journal":{"name":"Journal of the Peripheral Nervous System","volume":"28 4","pages":"642-650"},"PeriodicalIF":3.2000,"publicationDate":"2023-09-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/jns.12598","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the Peripheral Nervous System","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/jns.12598","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background and Aims
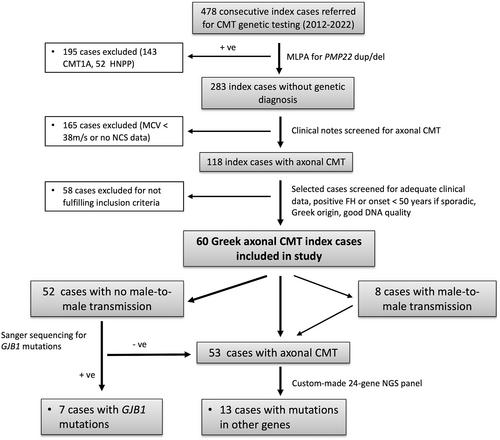
Axonal forms of Charcot-Marie-Tooth disease (CMT) are classified as CMT2, distal hereditary motor neuropathy (dHMN) or hereditary sensory neuropathy (HSN) and can be caused by mutations in over 100 genes. We presently aimed to investigate for the first time the genetic landscape of axonal CMT in the Greek population.
Methods
Sixty index patients with CMT2, dHMN or HSN were screened by a combination of Sanger sequencing (GJB1) and next-generation sequencing custom-made gene panel covering 24 commonly mutated genes in axonal CMT.
Results
Overall, 20 variants classified as pathogenic or likely pathogenic were identified in heterozygous state in 20 index cases, representing 33.3% of the cohort. Of these, 14 were known pathogenic/likely pathogenic and six were designated as such according to ACMG classification, after in silico evaluation, testing for familial segregation and further literature review. The most frequently involved genes were GJB1 (11.7%), MPZ (5%) and MFN2 (5%), followed by DNM2 (3.3%) and LRSAM1 (3.3%). Single cases were identified with mutations in BSCL2, HSPB1 and GDAP1.
Interpretation
A wide phenotypic variability in terms of severity and age of onset was noted. Given the limited number of genes tested, the diagnostic yield of the present panel compares favourably with studies in other European populations. Our study delineates the genetic and phenotypic variability of inherited axonal neuropathies in the Greek population and contributes to the pathogenicity characterization of further variants linked to axonal neuropathies.
期刊介绍:
The Journal of the Peripheral Nervous System is the official journal of the Peripheral Nerve Society. Founded in 1996, it is the scientific journal of choice for clinicians, clinical scientists and basic neuroscientists interested in all aspects of biology and clinical research of peripheral nervous system disorders.
The Journal of the Peripheral Nervous System is a peer-reviewed journal that publishes high quality articles on cell and molecular biology, genomics, neuropathic pain, clinical research, trials, and unique case reports on inherited and acquired peripheral neuropathies.
Original articles are organized according to the topic in one of four specific areas: Mechanisms of Disease, Genetics, Clinical Research, and Clinical Trials.
The journal also publishes regular review papers on hot topics and Special Issues on basic, clinical, or assembled research in the field of peripheral nervous system disorders. Authors interested in contributing a review-type article or a Special Issue should contact the Editorial Office to discuss the scope of the proposed article with the Editor-in-Chief.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
