{"title":"Biphasic function of GSK3β in gefitinib‑resistant NSCLC with or without EGFR mutations.","authors":"Junzhe Li, Xiayu Wu, Xiang-Bo Ji, Changhao He, Shijie Xu, Xianhua Xu","doi":"10.3892/etm.2023.12187","DOIUrl":null,"url":null,"abstract":"<p><p>Epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs), such as gefitinib, are effective in the treatment of non-small cell lung cancer (NSCLC) harboring EGFR mutations. However, the mechanism underlying acquired resistance to EGFR-TKIs remains largely unknown. Therefore, the present study generated gefitinib-resistant PC-9 (PC-9G) cells, which were revealed to be more resistant to gefitinib-induced reductions in proliferation, migration and invasion, and increases in apoptosis, and had no detectable EGFR mutations compared with the control PC-9 cell line. In addition, the present study performed genome-wide transcriptomic analysis of differentially expressed genes between PC-9 and PC-9G cell lines. Cell proliferation, colony formation, invasion, migration and flow cytometry analyses were also performed. The genome-wide transcriptomic analysis revealed that glycogen synthase kinase 3β (GSK3β) was downregulated in PC-9G cells compared with that in PC-9 cells. Furthermore, GSK3β overexpression increased the proliferation, migration and invasion of PC-9 and H1975 gefitinib-resistant cells. Conversely, overexpression of GSK3β suppressed the proliferation, migration and invasion of PC-9G cells. Furthermore, AKT inhibition reduced the proliferation, migration and invasion, and induced the apoptosis of PC-9, PC-9G and H1975 cells, the effects of which were reversed following AKT activation; notably, the tumor suppressor function of GSK3β was inconsistent with the tumor promotor role of the AKT pathway in PC-9G cells without EGFR mutation. The present study may provide novel insights into the distinctive role of GSK3β in gefitinib-resistant NSCLC with or without EGFR mutations, suggesting that a more detailed investigation on GSK3β as a therapeutic target for gefitinib-resistant NSCLC may be warranted.</p>","PeriodicalId":94002,"journal":{"name":"Experimental and therapeutic medicine","volume":"26 4","pages":"488"},"PeriodicalIF":2.3000,"publicationDate":"2023-09-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/75/01/etm-26-04-12187.PMC10515113.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Experimental and therapeutic medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3892/etm.2023.12187","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/10/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
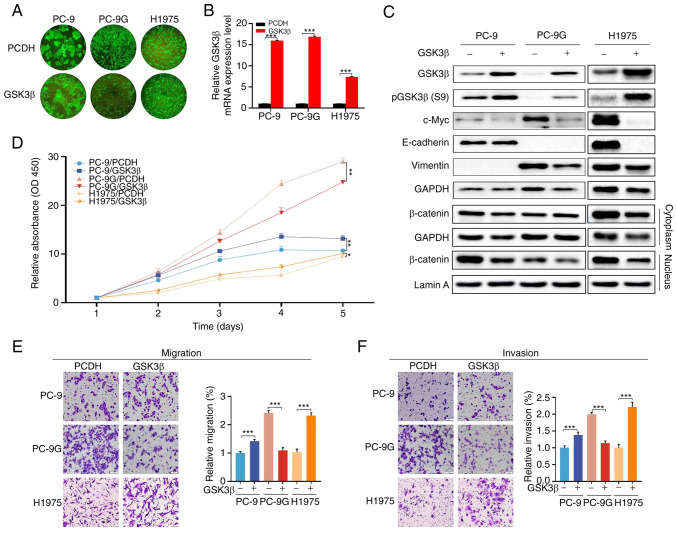
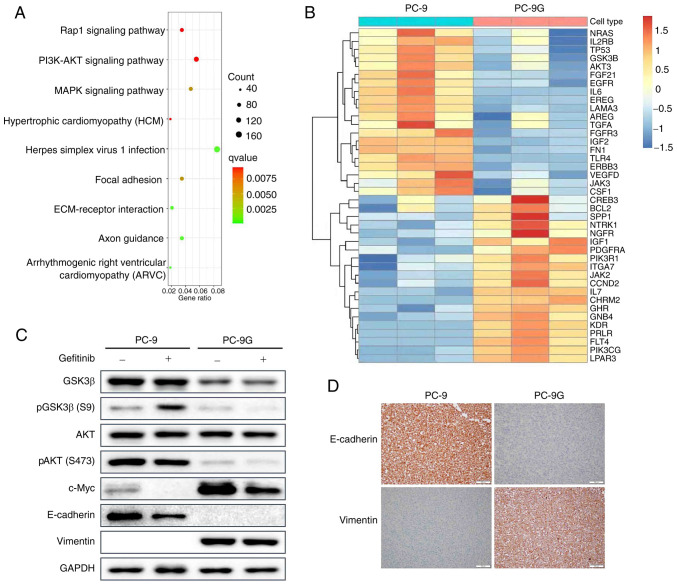
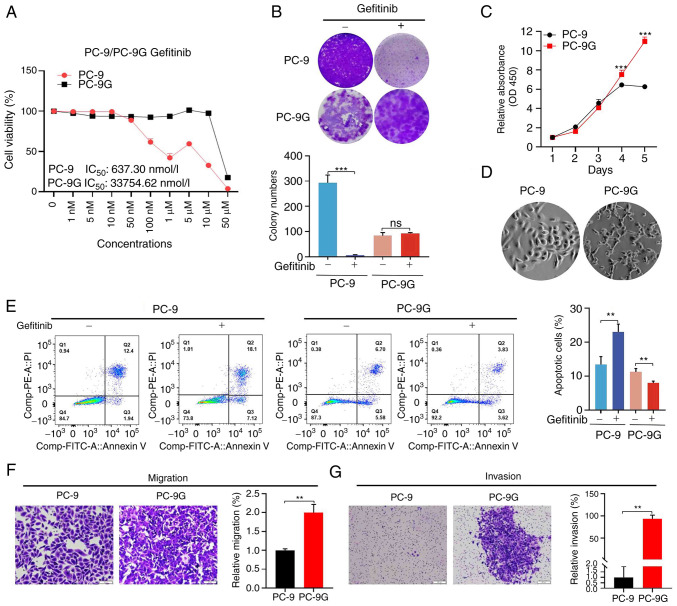
Epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs), such as gefitinib, are effective in the treatment of non-small cell lung cancer (NSCLC) harboring EGFR mutations. However, the mechanism underlying acquired resistance to EGFR-TKIs remains largely unknown. Therefore, the present study generated gefitinib-resistant PC-9 (PC-9G) cells, which were revealed to be more resistant to gefitinib-induced reductions in proliferation, migration and invasion, and increases in apoptosis, and had no detectable EGFR mutations compared with the control PC-9 cell line. In addition, the present study performed genome-wide transcriptomic analysis of differentially expressed genes between PC-9 and PC-9G cell lines. Cell proliferation, colony formation, invasion, migration and flow cytometry analyses were also performed. The genome-wide transcriptomic analysis revealed that glycogen synthase kinase 3β (GSK3β) was downregulated in PC-9G cells compared with that in PC-9 cells. Furthermore, GSK3β overexpression increased the proliferation, migration and invasion of PC-9 and H1975 gefitinib-resistant cells. Conversely, overexpression of GSK3β suppressed the proliferation, migration and invasion of PC-9G cells. Furthermore, AKT inhibition reduced the proliferation, migration and invasion, and induced the apoptosis of PC-9, PC-9G and H1975 cells, the effects of which were reversed following AKT activation; notably, the tumor suppressor function of GSK3β was inconsistent with the tumor promotor role of the AKT pathway in PC-9G cells without EGFR mutation. The present study may provide novel insights into the distinctive role of GSK3β in gefitinib-resistant NSCLC with or without EGFR mutations, suggesting that a more detailed investigation on GSK3β as a therapeutic target for gefitinib-resistant NSCLC may be warranted.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
