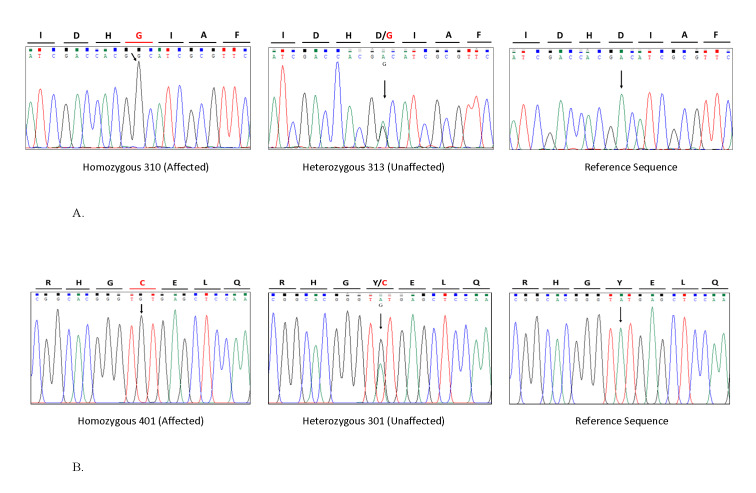
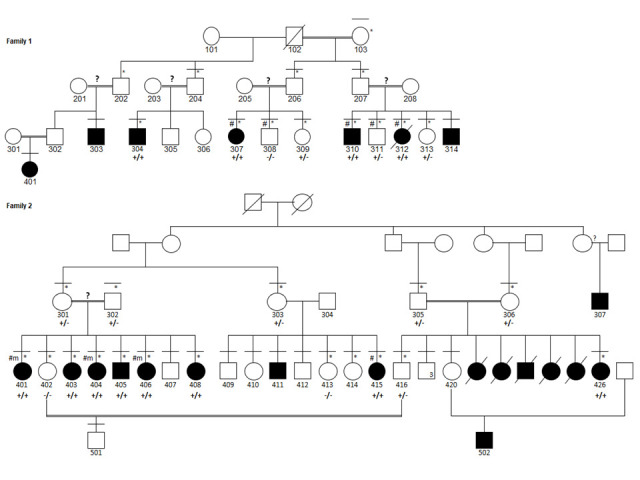
Homozygous Mutations in Thyroid Peroxidase (TPO) in Hypothyroidism with Intellectual Disability, Developmental Delay, and Hearing and Ocular Anomalies in Two Families: Severe Manifestation of Untreated TPO-deficiency Poses a Diagnostic Dilemma.
Syeda Farwa Naqvi, Esra Yıldız-Bölükbaşı, Muhammad Afzal, Gökhan Nalbant, Sara Mumtaz, Aslıhan Tolun, Sajid Malik
{"title":"Homozygous Mutations in Thyroid Peroxidase (TPO) in Hypothyroidism with Intellectual Disability, Developmental Delay, and Hearing and Ocular Anomalies in Two Families: Severe Manifestation of Untreated TPO-deficiency Poses a Diagnostic Dilemma.","authors":"Syeda Farwa Naqvi, Esra Yıldız-Bölükbaşı, Muhammad Afzal, Gökhan Nalbant, Sara Mumtaz, Aslıhan Tolun, Sajid Malik","doi":"10.59249/SSRG6507","DOIUrl":null,"url":null,"abstract":"<p><p>Intellectual disability (ID) involves compromised intellectual, learning and cognitive skills, and behavioral capabilities with reduced psychomotor skills. One of the preventable causes of ID is congenital hypothyroidism (CH), which may be due to biallelic mutations in <i>thyroid peroxidase</i> (<i>TPO</i>). In low- and middle-income countries with no newborn screening programs, CH poses a great risk of ID and long-term morbidity. We report two large Pakistani families with a total of 16 patients afflicted with CH. Detailed clinical and behavioral assessments, SNP-based homozygosity mapping, linkage analysis, and exome sequencing were performed. Initially, affected individuals were referred as suffering ID (in 11 of 16 patients) and developmental delay (in 14). Secondary/associated features were verbal apraxia (in 13), goiter (in 12), short stature (in 11), limb hypotonia (in 14), no pubertal onset (five of 10 of age ≥14 years), high myopia (in eight), muscle cramps (in six), and in some, variable microcephaly and enuresis/encopresis, fits, chronic fatigue, and other behavioral symptoms, which are not characteristics of CH. Molecular genetic analyses led to the discovery of homozygous variants in <i>TPO</i>: novel missense variant c.719A>G (p.Asp240Gly) in family 1 and rare c.2315A>G (p.Tyr772Cys) in family 2. In low-resource countries where neonatal screening programs do not include a CH test, the burden of neurodevelopmental disorders is likely to be increased due to untreated CH. Secondly, in the background of the high prevalence of recessive disorders due to high parental consanguinity, the severe manifestation of <i>TPO</i>-deficiency mimics a wide range of neurological and other presentations posing a diagnostic dilemma.</p>","PeriodicalId":48617,"journal":{"name":"Yale Journal of Biology and Medicine","volume":"96 3","pages":"347-365"},"PeriodicalIF":3.9000,"publicationDate":"2023-09-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/ac/03/yjbm_96_3_347.PMC10524819.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Yale Journal of Biology and Medicine","FirstCategoryId":"5","ListUrlMain":"https://doi.org/10.59249/SSRG6507","RegionNum":3,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Intellectual disability (ID) involves compromised intellectual, learning and cognitive skills, and behavioral capabilities with reduced psychomotor skills. One of the preventable causes of ID is congenital hypothyroidism (CH), which may be due to biallelic mutations in thyroid peroxidase (TPO). In low- and middle-income countries with no newborn screening programs, CH poses a great risk of ID and long-term morbidity. We report two large Pakistani families with a total of 16 patients afflicted with CH. Detailed clinical and behavioral assessments, SNP-based homozygosity mapping, linkage analysis, and exome sequencing were performed. Initially, affected individuals were referred as suffering ID (in 11 of 16 patients) and developmental delay (in 14). Secondary/associated features were verbal apraxia (in 13), goiter (in 12), short stature (in 11), limb hypotonia (in 14), no pubertal onset (five of 10 of age ≥14 years), high myopia (in eight), muscle cramps (in six), and in some, variable microcephaly and enuresis/encopresis, fits, chronic fatigue, and other behavioral symptoms, which are not characteristics of CH. Molecular genetic analyses led to the discovery of homozygous variants in TPO: novel missense variant c.719A>G (p.Asp240Gly) in family 1 and rare c.2315A>G (p.Tyr772Cys) in family 2. In low-resource countries where neonatal screening programs do not include a CH test, the burden of neurodevelopmental disorders is likely to be increased due to untreated CH. Secondly, in the background of the high prevalence of recessive disorders due to high parental consanguinity, the severe manifestation of TPO-deficiency mimics a wide range of neurological and other presentations posing a diagnostic dilemma.
期刊介绍:
The Yale Journal of Biology and Medicine (YJBM) is a graduate and medical student-run, peer-reviewed, open-access journal dedicated to the publication of original research articles, scientific reviews, articles on medical history, personal perspectives on medicine, policy analyses, case reports, and symposia related to biomedical matters. YJBM is published quarterly and aims to publish articles of interest to both physicians and scientists. YJBM is and has been an internationally distributed journal with a long history of landmark articles. Our contributors feature a notable list of philosophers, statesmen, scientists, and physicians, including Ernst Cassirer, Harvey Cushing, Rene Dubos, Edward Kennedy, Donald Seldin, and Jack Strominger. Our Editorial Board consists of students and faculty members from Yale School of Medicine and Yale University Graduate School of Arts & Sciences. All manuscripts submitted to YJBM are first evaluated on the basis of scientific quality, originality, appropriateness, contribution to the field, and style. Suitable manuscripts are then subject to rigorous, fair, and rapid peer review.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
