Saumya Maheshwari, Gabriela Vilema-Enríquez, Richard Wade-Martins
{"title":"Patient-derived iPSC models of Friedreich ataxia: a new frontier for understanding disease mechanisms and therapeutic application.","authors":"Saumya Maheshwari, Gabriela Vilema-Enríquez, Richard Wade-Martins","doi":"10.1186/s40035-023-00376-8","DOIUrl":null,"url":null,"abstract":"<p><p>Friedreich ataxia (FRDA) is a rare genetic multisystem disorder caused by a pathological GAA trinucleotide repeat expansion in the FXN gene. The numerous drawbacks of historical cellular and rodent models of FRDA have caused difficulty in performing effective mechanistic and translational studies to investigate the disease. The recent discovery and subsequent development of induced pluripotent stem cell (iPSC) technology provides an exciting platform to enable enhanced disease modelling for studies of rare genetic diseases. Utilising iPSCs, researchers have created phenotypically relevant and previously inaccessible cellular models of FRDA. These models enable studies of the molecular mechanisms underlying GAA-induced pathology, as well as providing an exciting tool for the screening and testing of novel disease-modifying therapies. This review explores how the use of iPSCs to study FRDA has developed over the past decade, as well as discussing the enormous therapeutic potentials of iPSC-derived models, their current limitations and their future direction within the field of FRDA research.</p>","PeriodicalId":23269,"journal":{"name":"Translational Neurodegeneration","volume":"12 1","pages":"45"},"PeriodicalIF":15.2000,"publicationDate":"2023-09-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10510273/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Translational Neurodegeneration","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s40035-023-00376-8","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
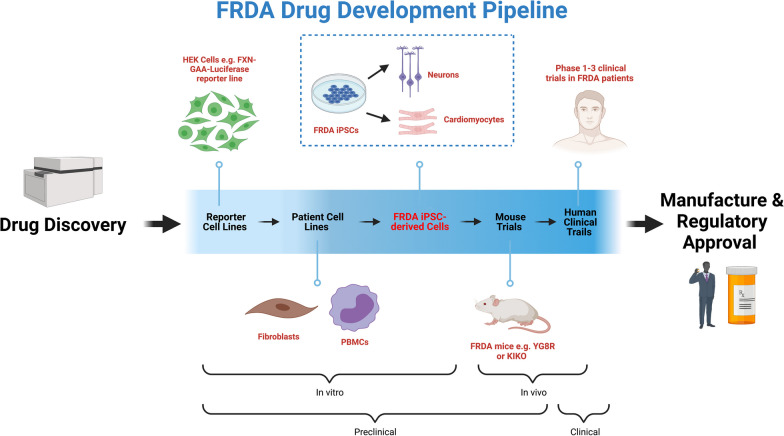
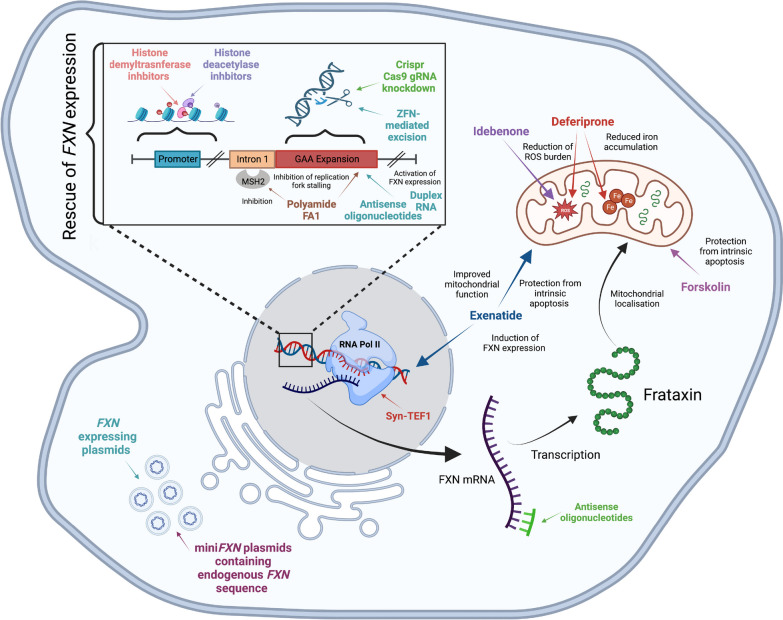
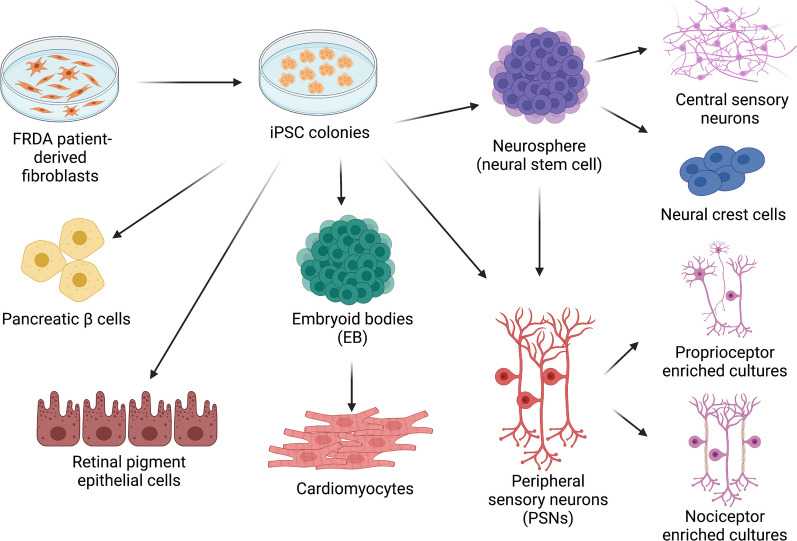
Friedreich ataxia (FRDA) is a rare genetic multisystem disorder caused by a pathological GAA trinucleotide repeat expansion in the FXN gene. The numerous drawbacks of historical cellular and rodent models of FRDA have caused difficulty in performing effective mechanistic and translational studies to investigate the disease. The recent discovery and subsequent development of induced pluripotent stem cell (iPSC) technology provides an exciting platform to enable enhanced disease modelling for studies of rare genetic diseases. Utilising iPSCs, researchers have created phenotypically relevant and previously inaccessible cellular models of FRDA. These models enable studies of the molecular mechanisms underlying GAA-induced pathology, as well as providing an exciting tool for the screening and testing of novel disease-modifying therapies. This review explores how the use of iPSCs to study FRDA has developed over the past decade, as well as discussing the enormous therapeutic potentials of iPSC-derived models, their current limitations and their future direction within the field of FRDA research.
期刊介绍:
Translational Neurodegeneration, an open-access, peer-reviewed journal, addresses all aspects of neurodegenerative diseases. It serves as a prominent platform for research, therapeutics, and education, fostering discussions and insights across basic, translational, and clinical research domains. Covering Parkinson's disease, Alzheimer's disease, and other neurodegenerative conditions, it welcomes contributions on epidemiology, pathogenesis, diagnosis, prevention, drug development, rehabilitation, and drug delivery. Scientists, clinicians, and physician-scientists are encouraged to share their work in this specialized journal tailored to their fields.


 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
