Robert Kleyner, Nathaniel Ung, Mohammad Arif, Elaine Marchi, Karen Amble, Maureen Gavin, Ricardo Madrid, Gholson Lyon
{"title":"<i>ITPR1</i>-associated spinocerebellar ataxia with craniofacial features-additional evidence for germline mosaicism.","authors":"Robert Kleyner, Nathaniel Ung, Mohammad Arif, Elaine Marchi, Karen Amble, Maureen Gavin, Ricardo Madrid, Gholson Lyon","doi":"10.1101/mcs.a006303","DOIUrl":null,"url":null,"abstract":"<p><p>Inositol 1,4,5-triphosphate receptor type 1 (<i>ITPR1</i>) is an endoplasmic reticulum-bound intracellular inositol triphosphate receptor involved in the regulation of intracellular calcium. Pathogenic variants in <i>ITPR1</i> are associated with spinocerebellar ataxia (SCA) types 15/16 and 29 and have recently been implicated in a facial microsomia syndrome. In this report, we present a family with three affected individuals found to have a heterozygous missense c.800C > T (predicted p.Thr267Met) who present clinically with a SCA29-like syndrome. All three individuals presented with varying degrees of ataxia, developmental delay, and apparent intellectual disability, as well as craniofacial involvement-an uncommon finding in patients with SCA29. The variant was identified using clinical exome sequencing and validated with Sanger sequencing. It is presumed to be inherited via parental germline mosaicism. We present our findings to provide additional evidence for germline mosaic inheritance of SCA29, as well as to expand the clinical phenotype of the syndrome.</p>","PeriodicalId":10360,"journal":{"name":"Cold Spring Harbor Molecular Case Studies","volume":" ","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2024-01-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10815276/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cold Spring Harbor Molecular Case Studies","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/mcs.a006303","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/12/1 0:00:00","PubModel":"Print","JCR":"Q3","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
Abstract
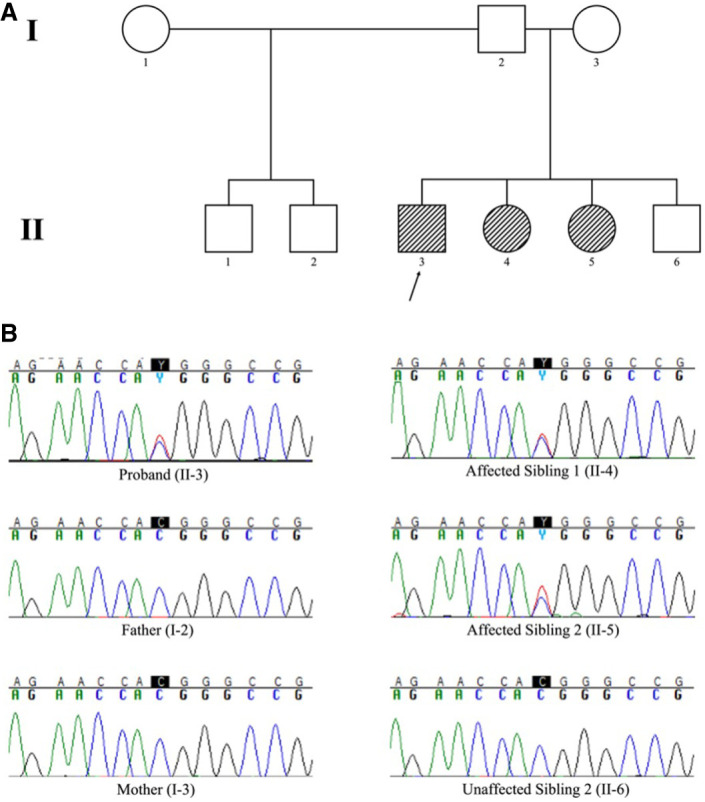
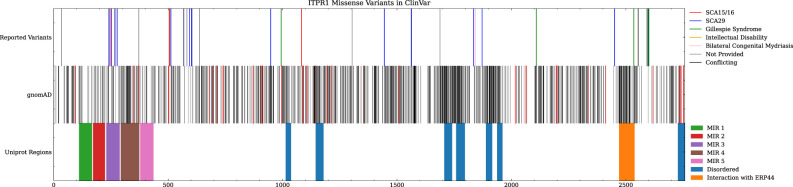
Inositol 1,4,5-triphosphate receptor type 1 (ITPR1) is an endoplasmic reticulum-bound intracellular inositol triphosphate receptor involved in the regulation of intracellular calcium. Pathogenic variants in ITPR1 are associated with spinocerebellar ataxia (SCA) types 15/16 and 29 and have recently been implicated in a facial microsomia syndrome. In this report, we present a family with three affected individuals found to have a heterozygous missense c.800C > T (predicted p.Thr267Met) who present clinically with a SCA29-like syndrome. All three individuals presented with varying degrees of ataxia, developmental delay, and apparent intellectual disability, as well as craniofacial involvement-an uncommon finding in patients with SCA29. The variant was identified using clinical exome sequencing and validated with Sanger sequencing. It is presumed to be inherited via parental germline mosaicism. We present our findings to provide additional evidence for germline mosaic inheritance of SCA29, as well as to expand the clinical phenotype of the syndrome.
期刊介绍:
Cold Spring Harbor Molecular Case Studies is an open-access, peer-reviewed, international journal in the field of precision medicine. Articles in the journal present genomic and molecular analyses of individuals or cohorts alongside their clinical presentations and phenotypic information. The journal''s purpose is to rapidly share insights into disease development and treatment gained by application of genomics, proteomics, metabolomics, biomarker analysis, and other approaches. The journal covers the fields of cancer, complex diseases, monogenic disorders, neurological conditions, orphan diseases, infectious disease, gene therapy, and pharmacogenomics. It has a rapid peer-review process that is based on technical evaluation of the analyses performed, not the novelty of findings, and offers a swift, clear path to publication. The journal publishes: Research Reports presenting detailed case studies of individuals and small cohorts, Research Articles describing more extensive work using larger cohorts and/or functional analyses, Rapid Communications presenting the discovery of a novel variant and/or novel phenotype associated with a known disease gene, Rapid Cancer Communications presenting the discovery of a novel variant or combination of variants in a cancer type, Variant Discrepancy Resolution describing efforts to resolve differences or update variant interpretations in ClinVar through case-level data sharing, Follow-up Reports linked to previous observations, Plus Review Articles, Editorials, and Position Statements on best practices for research in precision medicine.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
