Adarsh Arun, Dr. Zhen Guo, Dr. Simon Sung, Prof. Alexei A. Lapkin
{"title":"Reaction Impurity Prediction using a Data Mining Approach**","authors":"Adarsh Arun, Dr. Zhen Guo, Dr. Simon Sung, Prof. Alexei A. Lapkin","doi":"10.1002/cmtd.202200062","DOIUrl":null,"url":null,"abstract":"<p>Automated prediction of reaction impurities is useful in early-stage reaction development, synthesis planning and optimization. Existing reaction predictors are catered towards <i>main</i> product prediction, and are often black-box, making it difficult to troubleshoot erroneous outcomes. This work aims to present an automated, interpretable impurity prediction workflow based on data mining large chemical reaction databases. A 14-step workflow was implemented in Python and RDKit using Reaxys® data. Evaluation of potential chemical reactions between functional groups present in the same reaction environment in the user-supplied query species can be accurately performed by directly mining the Reaxys® database for similar or ‘analogue’ reactions involving these functional groups. Reaction templates can then be extracted from analogue reactions and applied to the relevant species in the original query to return impurities and transformations of interest. Three proof-of-concept case studies (paracetamol, agomelatine and lersivirine) were conducted, with the workflow correctly suggesting impurities within the top two outcomes. At all stages, suggested impurities can be traced back to the originating template and analogue reaction in the literature, allowing for closer inspection and user validation. Ultimately, this work could be useful as a benchmark for more sophisticated algorithms or models since it is interpretable, as opposed to purely black-box solutions.</p>","PeriodicalId":72562,"journal":{"name":"Chemistry methods : new approaches to solving problems in chemistry","volume":"3 6","pages":""},"PeriodicalIF":3.6000,"publicationDate":"2023-03-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cmtd.202200062","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemistry methods : new approaches to solving problems in chemistry","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/cmtd.202200062","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 1
Abstract
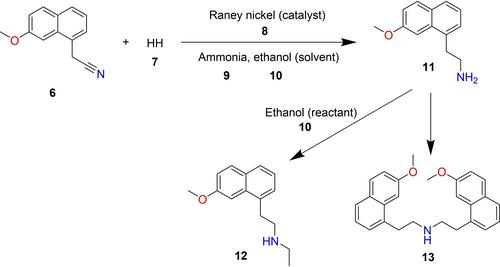
Automated prediction of reaction impurities is useful in early-stage reaction development, synthesis planning and optimization. Existing reaction predictors are catered towards main product prediction, and are often black-box, making it difficult to troubleshoot erroneous outcomes. This work aims to present an automated, interpretable impurity prediction workflow based on data mining large chemical reaction databases. A 14-step workflow was implemented in Python and RDKit using Reaxys® data. Evaluation of potential chemical reactions between functional groups present in the same reaction environment in the user-supplied query species can be accurately performed by directly mining the Reaxys® database for similar or ‘analogue’ reactions involving these functional groups. Reaction templates can then be extracted from analogue reactions and applied to the relevant species in the original query to return impurities and transformations of interest. Three proof-of-concept case studies (paracetamol, agomelatine and lersivirine) were conducted, with the workflow correctly suggesting impurities within the top two outcomes. At all stages, suggested impurities can be traced back to the originating template and analogue reaction in the literature, allowing for closer inspection and user validation. Ultimately, this work could be useful as a benchmark for more sophisticated algorithms or models since it is interpretable, as opposed to purely black-box solutions.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
