Douglas R. Nordli III, Erik Middlebrooks, Anteneh Feyissa
{"title":"Focal tonic absence seizure mischaracterized as periodic paralysis: A cautionary tale","authors":"Douglas R. Nordli III, Erik Middlebrooks, Anteneh Feyissa","doi":"10.1002/cns3.20013","DOIUrl":null,"url":null,"abstract":"<p>We highlight a patient whose focal epilepsy was misdiagnosed as periodic paralysis in order to better understand the diagnosis of periodic paralysis. In the evaluation of any mysterious spell, event capture and consideration of seizures are paramount.</p><p>This 24-year-old man with right hemiplegic spastic cerebral palsy presented to the epilepsy monitoring unit (EMU) for evaluation of episodic motor weakness that began at age ten. These events would last less than three minutes. He could speak and remained conscious during the spells. His parents reported that he appeared paralyzed during these episodes. He appeared “stuck” and had rigid tone and fixed posture. He underwent an extensive evaluation and was diagnosed with periodic paralysis (PP) and prescribed acetazolamide. In his 20s, he noted that an aura preceded these events. He experienced auditory hallucinations, a sensation of stomach acid in his throat, and fear, followed by an inability to move. He was evaluated for PP with electrolyte testing, genetics, and electromyography, all of which were normal.</p><p>Brain magnetic resonance imaging revealed a presumed perinatal cerebrovascular infarction in his left hemisphere (Figure 1), consistent with his findings of right spastic hemiplegic cerebral palsy.</p><p>During his EMU admission, a typical event was captured (Video 1). Based on these findings, his episodes were determined to be epilepsy and his treatment was modified to include valproic acid and discontinue acetazolamide. He was last seen three months after EMU admission and remained seizure-free on valproic acid monotherapy.</p><p>We describe a patient with focal epilepsy that was misdiagnosed as PP. He had never been admitted to an EMU. A thorough history and evaluation ultimately determined an alternative etiology of his episodes. Episodic weakness could be attributed to PP, nonepileptic events, metabolic derangements, cardiogenic syncope, or, as highlighted in this report, epileptic seizures.</p><p>The suspicion of PP was surprising, given the descriptions of his episodes. PP is a rare neuromuscular genetic disorder that can be generally divided into three categories: hyperkalemic, hypokalemic, and normokalemic. <i>CACNA1S</i>, <i>SCN4A</i>, and <i>KCNJ2</i> are the most common gene mutations.<span><sup>1</sup></span> Mutations of <i>SCN4A</i> most often present with hyperkalemic PP. <i>SCN4A</i> genes produce Na1.4 ion channels in muscle.<span><sup>2</sup></span> These ion channels allow an influx of sodium during the depolarization phase of an action potential.<span><sup>2</sup></span> The mutation leaves the sodium channel open for a prolonged period, thereby disabling further action potentials from generating. This shifts potassium outwards, resulting in elevated levels during attacks.<span><sup>2</sup></span> Hypokalemic periodic paralysis is often the result of <i>CACNA1S</i> mutations. These mutations affect Cav1.1 channels and cause a reduced ability for the muscle to depolarize.<span><sup>3</sup></span> Thus, in conditions of hypokalemia, the low extracellular ion concentration will cause the muscle to repolarize more readily, thereby disturbing calcium conductance and resulting in weakness.<span><sup>3</sup></span></p><p>PP is diagnosed through genetic testing, electromyography, or serum electrolyte levels during an attack.<span><sup>4</sup></span> Treatment of these attacks includes avoidance of triggers, diuretics, and carbonic anhydrase inhibitors.<span><sup>4</sup></span> Carbonic anhydrase inhibitors may be effective in PP.</p><p>Peculiar seizure semiologies, similar to our patient's, including tonic-absence seizure type, have been previously described.<span><sup>5</sup></span> The focal tonic arm stiffening is likely due to the involvement of the motor cortex. Given the quick bilateral nature of the motor seizure, the supplementary motor cortex is likely engaged early during the seizure. The slow spike and wave discharges afterward, with the associated absence, could have resulted from the involvement of the thalamo-cortical loop.</p><p>Seizure types that do not conform to traditional seizure classification schemes exist and can lead to misdiagnosis. Extreme detail in history taking is warranted in patients presenting with episodes of flaccid paralysis as small discrepancies often can help to tease apart the etiology of events. Indeed, even with a careful history collection, video-electroencephalography is warranted to capture the event and arrive at the correct diagnosis.</p><p><b>Douglas R. Nordli</b>: Data curation; writing – original draft. <b>Erik Middlebrooks</b>: Data curation; writing – review and editing. <b>Anteneh Feyissa</b>: Conceptualization; data curation; supervision; writing – original draft; writing – review and editing.</p><p>The authors declare no conflict of interest.</p>","PeriodicalId":72232,"journal":{"name":"Annals of the Child Neurology Society","volume":"1 1","pages":"82-84"},"PeriodicalIF":0.0000,"publicationDate":"2023-01-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cns3.20013","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Annals of the Child Neurology Society","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/cns3.20013","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
We highlight a patient whose focal epilepsy was misdiagnosed as periodic paralysis in order to better understand the diagnosis of periodic paralysis. In the evaluation of any mysterious spell, event capture and consideration of seizures are paramount.
This 24-year-old man with right hemiplegic spastic cerebral palsy presented to the epilepsy monitoring unit (EMU) for evaluation of episodic motor weakness that began at age ten. These events would last less than three minutes. He could speak and remained conscious during the spells. His parents reported that he appeared paralyzed during these episodes. He appeared “stuck” and had rigid tone and fixed posture. He underwent an extensive evaluation and was diagnosed with periodic paralysis (PP) and prescribed acetazolamide. In his 20s, he noted that an aura preceded these events. He experienced auditory hallucinations, a sensation of stomach acid in his throat, and fear, followed by an inability to move. He was evaluated for PP with electrolyte testing, genetics, and electromyography, all of which were normal.
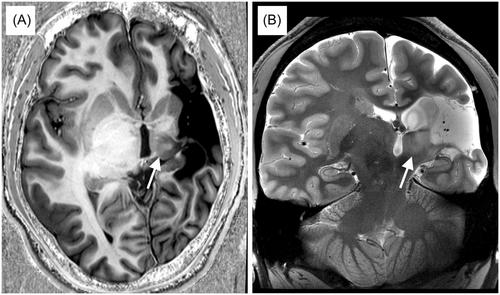
Brain magnetic resonance imaging revealed a presumed perinatal cerebrovascular infarction in his left hemisphere (Figure 1), consistent with his findings of right spastic hemiplegic cerebral palsy.
During his EMU admission, a typical event was captured (Video 1). Based on these findings, his episodes were determined to be epilepsy and his treatment was modified to include valproic acid and discontinue acetazolamide. He was last seen three months after EMU admission and remained seizure-free on valproic acid monotherapy.
We describe a patient with focal epilepsy that was misdiagnosed as PP. He had never been admitted to an EMU. A thorough history and evaluation ultimately determined an alternative etiology of his episodes. Episodic weakness could be attributed to PP, nonepileptic events, metabolic derangements, cardiogenic syncope, or, as highlighted in this report, epileptic seizures.
The suspicion of PP was surprising, given the descriptions of his episodes. PP is a rare neuromuscular genetic disorder that can be generally divided into three categories: hyperkalemic, hypokalemic, and normokalemic. CACNA1S, SCN4A, and KCNJ2 are the most common gene mutations.1 Mutations of SCN4A most often present with hyperkalemic PP. SCN4A genes produce Na1.4 ion channels in muscle.2 These ion channels allow an influx of sodium during the depolarization phase of an action potential.2 The mutation leaves the sodium channel open for a prolonged period, thereby disabling further action potentials from generating. This shifts potassium outwards, resulting in elevated levels during attacks.2 Hypokalemic periodic paralysis is often the result of CACNA1S mutations. These mutations affect Cav1.1 channels and cause a reduced ability for the muscle to depolarize.3 Thus, in conditions of hypokalemia, the low extracellular ion concentration will cause the muscle to repolarize more readily, thereby disturbing calcium conductance and resulting in weakness.3
PP is diagnosed through genetic testing, electromyography, or serum electrolyte levels during an attack.4 Treatment of these attacks includes avoidance of triggers, diuretics, and carbonic anhydrase inhibitors.4 Carbonic anhydrase inhibitors may be effective in PP.
Peculiar seizure semiologies, similar to our patient's, including tonic-absence seizure type, have been previously described.5 The focal tonic arm stiffening is likely due to the involvement of the motor cortex. Given the quick bilateral nature of the motor seizure, the supplementary motor cortex is likely engaged early during the seizure. The slow spike and wave discharges afterward, with the associated absence, could have resulted from the involvement of the thalamo-cortical loop.
Seizure types that do not conform to traditional seizure classification schemes exist and can lead to misdiagnosis. Extreme detail in history taking is warranted in patients presenting with episodes of flaccid paralysis as small discrepancies often can help to tease apart the etiology of events. Indeed, even with a careful history collection, video-electroencephalography is warranted to capture the event and arrive at the correct diagnosis.
Douglas R. Nordli: Data curation; writing – original draft. Erik Middlebrooks: Data curation; writing – review and editing. Anteneh Feyissa: Conceptualization; data curation; supervision; writing – original draft; writing – review and editing.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
