Andrew McKnight, Dong Si, Kamal Al Nasr, Andrey Chernikov, Nikos Chrisochoides, Jing He
{"title":"Estimating loop length from CryoEM images at medium resolutions","authors":"Andrew McKnight, Dong Si, Kamal Al Nasr, Andrey Chernikov, Nikos Chrisochoides, Jing He","doi":"10.1186/1472-6807-13-S1-S5","DOIUrl":null,"url":null,"abstract":"<p>De novo protein modeling approaches utilize 3-dimensional (3D) images derived from electron cryomicroscopy (CryoEM) experiments. The skeleton connecting two secondary structures such as <i>α</i>-helices represent the loop in the 3D image. The accuracy of the skeleton and of the detected secondary structures are critical in De novo modeling. It is important to measure the length along the skeleton accurately since the length can be used as a constraint in modeling the protein.</p><p>We have developed a novel computational geometric approach to derive a simplified curve in order to estimate the loop length along the skeleton. The method was tested using fifty simulated density images of helix-loop-helix segments of atomic structures and eighteen experimentally derived density data from Electron Microscopy Data Bank (EMDB). The test using simulated density maps shows that it is possible to estimate within 0.5? of the expected length for 48 of the 50 cases. The experiments, involving eighteen experimentally derived CryoEM images, show that twelve cases have error within 2?.</p><p>The tests using both simulated and experimentally derived images show that it is possible for our proposed method to estimate the loop length along the skeleton if the secondary structure elements, such as <i>α</i>-helices, can be detected accurately, and there is a continuous skeleton linking the <i>α</i>-helices.</p>","PeriodicalId":51240,"journal":{"name":"BMC Structural Biology","volume":"13 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2013-11-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1472-6807-13-S1-S5","citationCount":"7","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/1472-6807-13-S1-S5","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 7
Abstract
De novo protein modeling approaches utilize 3-dimensional (3D) images derived from electron cryomicroscopy (CryoEM) experiments. The skeleton connecting two secondary structures such as α-helices represent the loop in the 3D image. The accuracy of the skeleton and of the detected secondary structures are critical in De novo modeling. It is important to measure the length along the skeleton accurately since the length can be used as a constraint in modeling the protein.
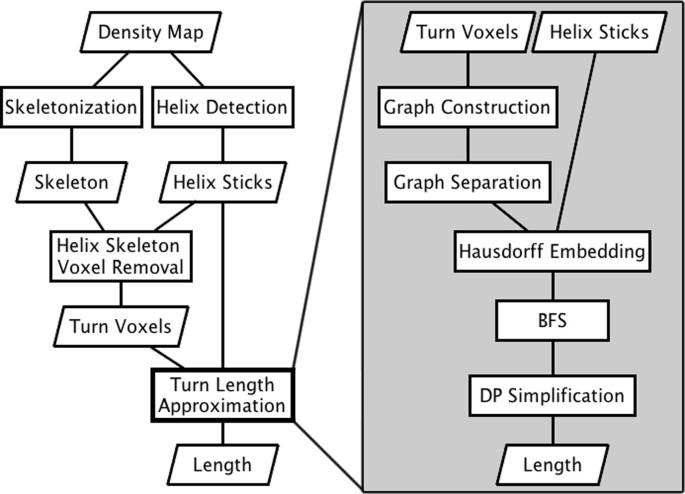
We have developed a novel computational geometric approach to derive a simplified curve in order to estimate the loop length along the skeleton. The method was tested using fifty simulated density images of helix-loop-helix segments of atomic structures and eighteen experimentally derived density data from Electron Microscopy Data Bank (EMDB). The test using simulated density maps shows that it is possible to estimate within 0.5? of the expected length for 48 of the 50 cases. The experiments, involving eighteen experimentally derived CryoEM images, show that twelve cases have error within 2?.
The tests using both simulated and experimentally derived images show that it is possible for our proposed method to estimate the loop length along the skeleton if the secondary structure elements, such as α-helices, can be detected accurately, and there is a continuous skeleton linking the α-helices.
期刊介绍:
BMC Structural Biology is an open access, peer-reviewed journal that considers articles on investigations into the structure of biological macromolecules, including solving structures, structural and functional analyses, and computational modeling.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
