Ibrahim Al-Bluwi, Marc Vaisset, Thierry Siméon, Juan Cortés
{"title":"Modeling protein conformational transitions by a combination of coarse-grained normal mode analysis and robotics-inspired methods","authors":"Ibrahim Al-Bluwi, Marc Vaisset, Thierry Siméon, Juan Cortés","doi":"10.1186/1472-6807-13-S1-S2","DOIUrl":null,"url":null,"abstract":"<p>Obtaining atomic-scale information about large-amplitude conformational transitions in proteins is a challenging problem for both experimental and computational methods. Such information is, however, important for understanding the mechanisms of interaction of many proteins.</p><p>This paper presents a computationally efficient approach, combining methods originating from robotics and computational biophysics, to model protein conformational transitions. The ability of normal mode analysis to predict directions of collective, large-amplitude motions is applied to bias the conformational exploration performed by a motion planning algorithm. To reduce the dimension of the problem, normal modes are computed for a coarse-grained elastic network model built on short fragments of three residues. Nevertheless, the validity of intermediate conformations is checked using the all-atom model, which is accurately reconstructed from the coarse-grained one using closed-form inverse kinematics.</p><p>Tests on a set of ten proteins demonstrate the ability of the method to model conformational transitions of proteins within a few hours of computing time on a single processor. These results also show that the computing time scales linearly with the protein size, independently of the protein topology. Further experiments on adenylate kinase show that main features of the transition between the open and closed conformations of this protein are well captured in the computed path.</p><p>The proposed method enables the simulation of large-amplitude conformational transitions in proteins using very few computational resources. The resulting paths are a first approximation that can directly provide important information on the molecular mechanisms involved in the conformational transition. This approximation can be subsequently refined and analyzed using state-of-the-art energy models and molecular modeling methods.</p>","PeriodicalId":51240,"journal":{"name":"BMC Structural Biology","volume":"13 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2013-11-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1472-6807-13-S1-S2","citationCount":"52","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/1472-6807-13-S1-S2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 52
Abstract
Obtaining atomic-scale information about large-amplitude conformational transitions in proteins is a challenging problem for both experimental and computational methods. Such information is, however, important for understanding the mechanisms of interaction of many proteins.
This paper presents a computationally efficient approach, combining methods originating from robotics and computational biophysics, to model protein conformational transitions. The ability of normal mode analysis to predict directions of collective, large-amplitude motions is applied to bias the conformational exploration performed by a motion planning algorithm. To reduce the dimension of the problem, normal modes are computed for a coarse-grained elastic network model built on short fragments of three residues. Nevertheless, the validity of intermediate conformations is checked using the all-atom model, which is accurately reconstructed from the coarse-grained one using closed-form inverse kinematics.
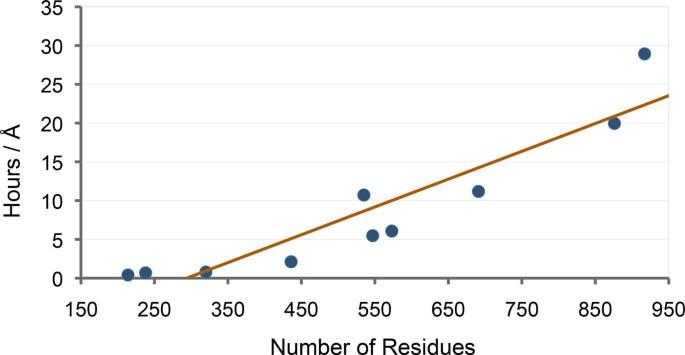
Tests on a set of ten proteins demonstrate the ability of the method to model conformational transitions of proteins within a few hours of computing time on a single processor. These results also show that the computing time scales linearly with the protein size, independently of the protein topology. Further experiments on adenylate kinase show that main features of the transition between the open and closed conformations of this protein are well captured in the computed path.
The proposed method enables the simulation of large-amplitude conformational transitions in proteins using very few computational resources. The resulting paths are a first approximation that can directly provide important information on the molecular mechanisms involved in the conformational transition. This approximation can be subsequently refined and analyzed using state-of-the-art energy models and molecular modeling methods.
期刊介绍:
BMC Structural Biology is an open access, peer-reviewed journal that considers articles on investigations into the structure of biological macromolecules, including solving structures, structural and functional analyses, and computational modeling.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
