{"title":"Four-body atomic potential for modeling protein-ligand binding affinity: application to enzyme-inhibitor binding energy prediction","authors":"Majid Masso","doi":"10.1186/1472-6807-13-S1-S1","DOIUrl":null,"url":null,"abstract":"<p>Models that are capable of reliably predicting binding affinities for protein-ligand complexes play an important role the field of structure-guided drug design.</p><p>Here, we begin by applying the computational geometry technique of Delaunay tessellation to each set of atomic coordinates for over 1400 diverse macromolecular structures, for the purpose of deriving a four-body statistical potential that serves as a topological scoring function. Next, we identify a second, independent set of three hundred protein-ligand complexes, having both high-resolution structures and known dissociation constants. Two-thirds of these complexes are randomly selected to train a predictive model of binding affinity as follows: two tessellations are generated in each case, one for the entire complex and another strictly for the isolated protein without its bound ligand, and a topological score is computed for each tessellation with the four-body potential. Predicted protein-ligand binding affinity is then based on an empirically derived linear function of the difference between both topological scores, one that appropriately scales the value of this difference.</p><p>A comparison between experimental and calculated binding affinity values over the two hundred complexes reveals a Pearson's correlation coefficient of <i>r</i> = 0.79 with a standard error of <i>SE</i> = 1.98 kcal/mol. To validate the method, we similarly generated two tessellations for each of the remaining protein-ligand complexes, computed their topological scores and the difference between the two scores for each complex, and applied the previously derived linear transformation of this topological score difference to predict binding affinities. For these one hundred complexes, we again observe a correlation of <i>r</i> = 0.79 (<i>SE</i> = 1.93 kcal/mol) between known and calculated binding affinities. Applying our model to an independent test set of high-resolution structures for three hundred diverse enzyme-inhibitor complexes, each with an experimentally known inhibition constant, also yields a correlation of <i>r</i> = 0.79 (<i>SE</i> = 2.39 kcal/mol) between experimental and calculated binding energies.</p><p>Lastly, we generate predictions with our model on a diverse test set of one hundred protein-ligand complexes previously used to benchmark 15 related methods, and our correlation of <i>r</i> = 0.66 between the calculated and experimental binding energies for this dataset exceeds those of the other approaches. Compared with these related prediction methods, our approach stands out based on salient features that include the reliability of our model, combined with the rapidity of the generated predictions, which are less than one second for an average sized complex.</p>","PeriodicalId":51240,"journal":{"name":"BMC Structural Biology","volume":"13 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2013-11-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1472-6807-13-S1-S1","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/1472-6807-13-S1-S1","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 4
Abstract
Models that are capable of reliably predicting binding affinities for protein-ligand complexes play an important role the field of structure-guided drug design.
Here, we begin by applying the computational geometry technique of Delaunay tessellation to each set of atomic coordinates for over 1400 diverse macromolecular structures, for the purpose of deriving a four-body statistical potential that serves as a topological scoring function. Next, we identify a second, independent set of three hundred protein-ligand complexes, having both high-resolution structures and known dissociation constants. Two-thirds of these complexes are randomly selected to train a predictive model of binding affinity as follows: two tessellations are generated in each case, one for the entire complex and another strictly for the isolated protein without its bound ligand, and a topological score is computed for each tessellation with the four-body potential. Predicted protein-ligand binding affinity is then based on an empirically derived linear function of the difference between both topological scores, one that appropriately scales the value of this difference.
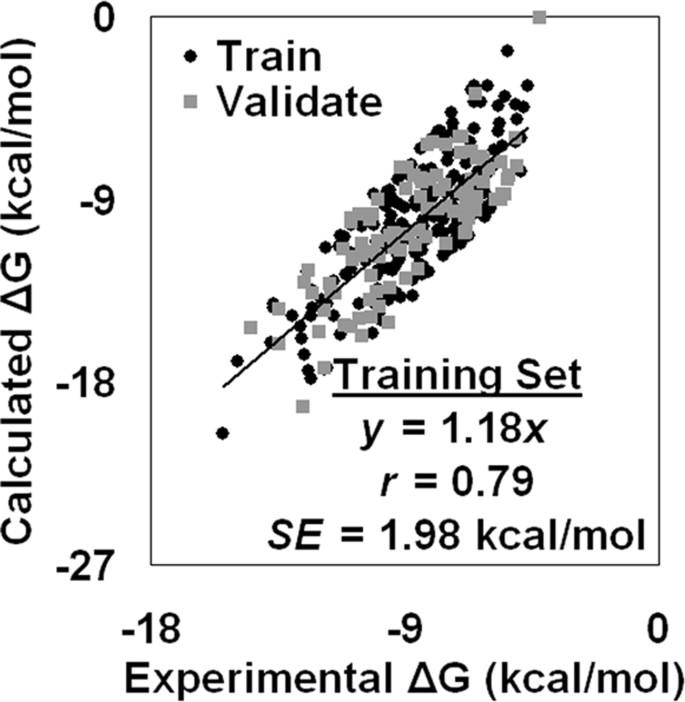
A comparison between experimental and calculated binding affinity values over the two hundred complexes reveals a Pearson's correlation coefficient of r = 0.79 with a standard error of SE = 1.98 kcal/mol. To validate the method, we similarly generated two tessellations for each of the remaining protein-ligand complexes, computed their topological scores and the difference between the two scores for each complex, and applied the previously derived linear transformation of this topological score difference to predict binding affinities. For these one hundred complexes, we again observe a correlation of r = 0.79 (SE = 1.93 kcal/mol) between known and calculated binding affinities. Applying our model to an independent test set of high-resolution structures for three hundred diverse enzyme-inhibitor complexes, each with an experimentally known inhibition constant, also yields a correlation of r = 0.79 (SE = 2.39 kcal/mol) between experimental and calculated binding energies.
Lastly, we generate predictions with our model on a diverse test set of one hundred protein-ligand complexes previously used to benchmark 15 related methods, and our correlation of r = 0.66 between the calculated and experimental binding energies for this dataset exceeds those of the other approaches. Compared with these related prediction methods, our approach stands out based on salient features that include the reliability of our model, combined with the rapidity of the generated predictions, which are less than one second for an average sized complex.
期刊介绍:
BMC Structural Biology is an open access, peer-reviewed journal that considers articles on investigations into the structure of biological macromolecules, including solving structures, structural and functional analyses, and computational modeling.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
