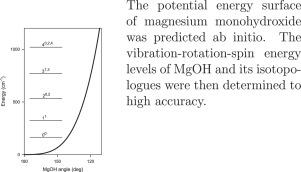
{"title":"Ab initio potential energy surface and vibration-rotation energy levels of magnesium monohydroxide revisited","authors":"Jacek Koput","doi":"10.1016/j.jms.2023.111805","DOIUrl":null,"url":null,"abstract":"<div><p><span>The accurate potential energy surface of magnesium monohydroxide, MgOH, in its ground electronic state </span><span><math><mrow><mover><mrow><mi>X</mi></mrow><mrow><mo>˜</mo></mrow></mover><msup><mrow><mspace></mspace></mrow><mrow><mn>2</mn></mrow></msup><msup><mrow><mi>Σ</mi></mrow><mrow><mo>+</mo></mrow></msup></mrow></math></span><span> has been determined from ab initio calculations<span><span> using the coupled-cluster approach in conjunction with the correlation-consistent basis sets up to septuple-zeta quality. The core-electron correlation, higher-order electron correlation, scalar relativistic, and adiabatic effects were taken into account. The equilibrium configuration of the MgOH molecule was confirmed to be linear, although with the bending </span>potential energy function being very flat near the minimum. The vibration–rotation-spin energy levels of the MgOH, MgOD, </span></span><sup>25</sup>MgOH, and <sup>26</sup>MgOH isotopologues were predicted using a variational approach. The spectroscopic constants of these isotopologues were determined to high accuracy.</p></div>","PeriodicalId":16367,"journal":{"name":"Journal of Molecular Spectroscopy","volume":"395 ","pages":"Article 111805"},"PeriodicalIF":1.3000,"publicationDate":"2023-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Spectroscopy","FirstCategoryId":"101","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S002228522300070X","RegionNum":4,"RegionCategory":"物理与天体物理","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"PHYSICS, ATOMIC, MOLECULAR & CHEMICAL","Score":null,"Total":0}
引用次数: 1
Abstract
The accurate potential energy surface of magnesium monohydroxide, MgOH, in its ground electronic state has been determined from ab initio calculations using the coupled-cluster approach in conjunction with the correlation-consistent basis sets up to septuple-zeta quality. The core-electron correlation, higher-order electron correlation, scalar relativistic, and adiabatic effects were taken into account. The equilibrium configuration of the MgOH molecule was confirmed to be linear, although with the bending potential energy function being very flat near the minimum. The vibration–rotation-spin energy levels of the MgOH, MgOD, 25MgOH, and 26MgOH isotopologues were predicted using a variational approach. The spectroscopic constants of these isotopologues were determined to high accuracy.
期刊介绍:
The Journal of Molecular Spectroscopy presents experimental and theoretical articles on all subjects relevant to molecular spectroscopy and its modern applications. An international medium for the publication of some of the most significant research in the field, the Journal of Molecular Spectroscopy is an invaluable resource for astrophysicists, chemists, physicists, engineers, and others involved in molecular spectroscopy research and practice.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
