{"title":"Modeling of the OX1R–orexin-A complex suggests two alternative binding modes","authors":"Lasse Karhu, Ainoleena Turku, Henri Xhaard","doi":"10.1186/s12900-015-0036-2","DOIUrl":null,"url":null,"abstract":"<p>Interactions between the orexin peptides and their cognate OX<sub>1</sub> and OX<sub>2</sub> receptors remain poorly characterized. Site-directed mutagenesis studies on orexin peptides and receptors have indicated amino acids important for ligand binding and receptor activation. However, a better understanding of specific pairwise interactions would benefit small molecule discovery.</p><p>We constructed a set of three-dimensional models of the orexin 1 receptor based on the 3D-structures of the orexin 2 receptor (released while this manuscript was under review), neurotensin receptor 1 and chemokine receptor CXCR4, conducted an exhaustive docking of orexin-A<sub>16–33</sub> peptide fragment with ZDOCK and RDOCK, and analyzed a total of 4301 complexes through multidimensional scaling and clustering. The best docking poses reveal two alternative binding modes, where the C-terminus of the peptide lies deep in the binding pocket, on average about 5–6?? above Tyr<sup>6.48</sup> and close to Gln<sup>3.32</sup>. The binding modes differ in the about 100° rotation of the peptide; the peptide His26 faces either the receptor’s fifth transmembrane helix or the seventh helix. Both binding modes are well in line with previous mutation studies and partake in hydrogen bonding similar to suvorexant.</p><p>We present two binding modes for orexin-A into orexin 1 receptor, which help rationalize previous results from site-directed mutagenesis studies. The binding modes should serve small molecule discovery, and offer insights into the mechanism of receptor activation.</p>","PeriodicalId":498,"journal":{"name":"BMC Structural Biology","volume":"15 1","pages":""},"PeriodicalIF":2.2220,"publicationDate":"2015-05-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12900-015-0036-2","citationCount":"22","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/s12900-015-0036-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 22
Abstract
Interactions between the orexin peptides and their cognate OX1 and OX2 receptors remain poorly characterized. Site-directed mutagenesis studies on orexin peptides and receptors have indicated amino acids important for ligand binding and receptor activation. However, a better understanding of specific pairwise interactions would benefit small molecule discovery.
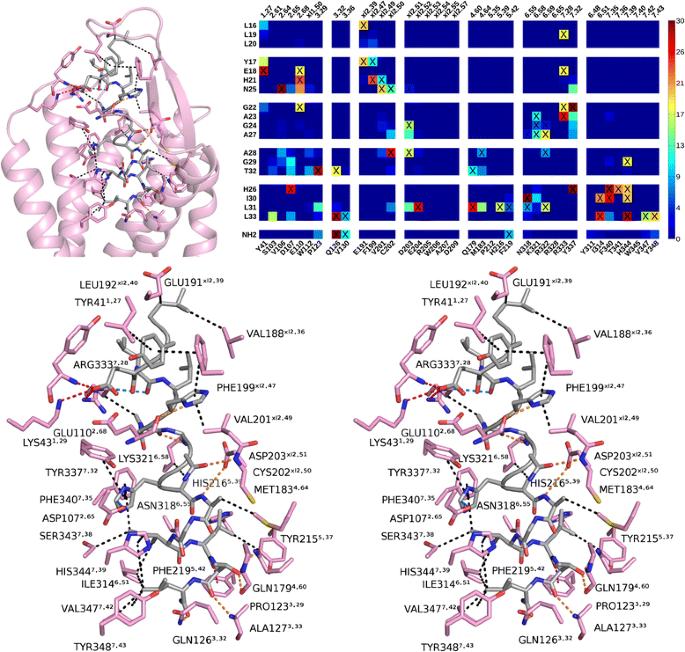
We constructed a set of three-dimensional models of the orexin 1 receptor based on the 3D-structures of the orexin 2 receptor (released while this manuscript was under review), neurotensin receptor 1 and chemokine receptor CXCR4, conducted an exhaustive docking of orexin-A16–33 peptide fragment with ZDOCK and RDOCK, and analyzed a total of 4301 complexes through multidimensional scaling and clustering. The best docking poses reveal two alternative binding modes, where the C-terminus of the peptide lies deep in the binding pocket, on average about 5–6?? above Tyr6.48 and close to Gln3.32. The binding modes differ in the about 100° rotation of the peptide; the peptide His26 faces either the receptor’s fifth transmembrane helix or the seventh helix. Both binding modes are well in line with previous mutation studies and partake in hydrogen bonding similar to suvorexant.
We present two binding modes for orexin-A into orexin 1 receptor, which help rationalize previous results from site-directed mutagenesis studies. The binding modes should serve small molecule discovery, and offer insights into the mechanism of receptor activation.
期刊介绍:
BMC Structural Biology is an open access, peer-reviewed journal that considers articles on investigations into the structure of biological macromolecules, including solving structures, structural and functional analyses, and computational modeling.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
