Rosetta Broker for membrane protein structure prediction: concentrative nucleoside transporter 3 and corticotropin-releasing factor receptor 1 test cases
{"title":"Rosetta Broker for membrane protein structure prediction: concentrative nucleoside transporter 3 and corticotropin-releasing factor receptor 1 test cases","authors":"Dorota Latek","doi":"10.1186/s12900-017-0078-8","DOIUrl":null,"url":null,"abstract":"<p>Membrane proteins are difficult targets for structure prediction due to the limited structural data deposited in Protein Data Bank. Most computational methods for membrane protein structure prediction are based on the comparative modeling. There are only few de novo methods targeting that distinct protein family. In this work an example of such de novo method was used to structurally and functionally characterize two representatives of distinct membrane proteins families of solute carrier transporters and G protein-coupled receptors. The well-known Rosetta program and one of its protocols named Broker was used in two test cases. The first case was de novo structure prediction of three N-terminal transmembrane helices of the human concentrative nucleoside transporter 3 (hCNT3) homotrimer belonging to the solute carrier 28 family of transporters (SLC28). The second case concerned the large scale refinement of transmembrane helices of a homology model of the corticotropin-releasing factor receptor 1 (CRFR1) belonging to the G protein-coupled receptors family.</p><p>The inward-facing model of the hCNT3 homotrimer was used to propose the functional impact of its single nucleotide polymorphisms. Additionally, the 100?ns molecular dynamics simulation of the unliganded hCNT3 model confirmed its validity and revealed mobility of the selected binding site and homotrimer interface residues. The large scale refinement of transmembrane helices of the CRFR1 homology model resulted in the significant improvement of its accuracy with respect to the crystal structure of CRFR1, especially in the binding site area. Consequently, the antagonist CP-376395 could be docked with Autodock VINA to the CRFR1 model without any steric clashes.</p><p>The presented work demonstrated that Rosetta Broker can be a versatile tool for solving various issues referring to protein biology. Two distinct examples of de novo membrane protein structure prediction presented here provided important insights into three major areas of protein biology. Namely, the dynamics of the inward-facing hCNT3 homotrimer system, the structural changes of the CRFR1 receptor upon the antagonist binding and finally, the role of single nucleotide polymorphisms in both, hCNT3 and CRFR1 proteins, were investigated.</p>","PeriodicalId":498,"journal":{"name":"BMC Structural Biology","volume":"17 1","pages":""},"PeriodicalIF":2.2220,"publicationDate":"2017-08-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12900-017-0078-8","citationCount":"6","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/s12900-017-0078-8","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 6
Abstract
Membrane proteins are difficult targets for structure prediction due to the limited structural data deposited in Protein Data Bank. Most computational methods for membrane protein structure prediction are based on the comparative modeling. There are only few de novo methods targeting that distinct protein family. In this work an example of such de novo method was used to structurally and functionally characterize two representatives of distinct membrane proteins families of solute carrier transporters and G protein-coupled receptors. The well-known Rosetta program and one of its protocols named Broker was used in two test cases. The first case was de novo structure prediction of three N-terminal transmembrane helices of the human concentrative nucleoside transporter 3 (hCNT3) homotrimer belonging to the solute carrier 28 family of transporters (SLC28). The second case concerned the large scale refinement of transmembrane helices of a homology model of the corticotropin-releasing factor receptor 1 (CRFR1) belonging to the G protein-coupled receptors family.
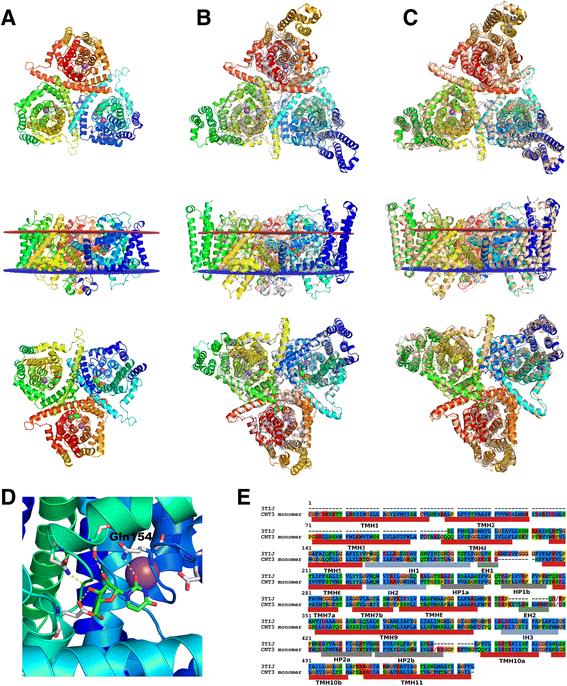
The inward-facing model of the hCNT3 homotrimer was used to propose the functional impact of its single nucleotide polymorphisms. Additionally, the 100?ns molecular dynamics simulation of the unliganded hCNT3 model confirmed its validity and revealed mobility of the selected binding site and homotrimer interface residues. The large scale refinement of transmembrane helices of the CRFR1 homology model resulted in the significant improvement of its accuracy with respect to the crystal structure of CRFR1, especially in the binding site area. Consequently, the antagonist CP-376395 could be docked with Autodock VINA to the CRFR1 model without any steric clashes.
The presented work demonstrated that Rosetta Broker can be a versatile tool for solving various issues referring to protein biology. Two distinct examples of de novo membrane protein structure prediction presented here provided important insights into three major areas of protein biology. Namely, the dynamics of the inward-facing hCNT3 homotrimer system, the structural changes of the CRFR1 receptor upon the antagonist binding and finally, the role of single nucleotide polymorphisms in both, hCNT3 and CRFR1 proteins, were investigated.
期刊介绍:
BMC Structural Biology is an open access, peer-reviewed journal that considers articles on investigations into the structure of biological macromolecules, including solving structures, structural and functional analyses, and computational modeling.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
