Mutational Patterns Observed in SARS-CoV-2 Genomes Sampled From Successive Epochs Delimited by Major Public Health Events in Ontario, Canada: Genomic Surveillance Study.
David Chen, Gurjit S Randhawa, Maximillian Pm Soltysiak, Camila Pe de Souza, Lila Kari, Shiva M Singh, Kathleen A Hill
{"title":"Mutational Patterns Observed in SARS-CoV-2 Genomes Sampled From Successive Epochs Delimited by Major Public Health Events in Ontario, Canada: Genomic Surveillance Study.","authors":"David Chen, Gurjit S Randhawa, Maximillian Pm Soltysiak, Camila Pe de Souza, Lila Kari, Shiva M Singh, Kathleen A Hill","doi":"10.2196/42243","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The emergence of SARS-CoV-2 variants with mutations associated with increased transmissibility and virulence is a public health concern in Ontario, Canada. Characterizing how the mutational patterns of the SARS-CoV-2 genome have changed over time can shed light on the driving factors, including selection for increased fitness and host immune response, that may contribute to the emergence of novel variants. Moreover, the study of SARS-CoV-2 in the microcosm of Ontario, Canada can reveal how different province-specific public health policies over time may be associated with observed mutational patterns as a model system.</p><p><strong>Objective: </strong>This study aimed to perform a comprehensive analysis of single base substitution (SBS) types, counts, and genomic locations observed in SARS-CoV-2 genomic sequences sampled in Ontario, Canada. Comparisons of mutational patterns were conducted between sequences sampled during 4 different epochs delimited by major public health events to track the evolution of the SARS-CoV-2 mutational landscape over 2 years.</p><p><strong>Methods: </strong>In total, 24,244 SARS-CoV-2 genomic sequences and associated metadata sampled in Ontario, Canada from January 1, 2020, to December 31, 2021, were retrieved from the Global Initiative on Sharing All Influenza Data database. Sequences were assigned to 4 epochs delimited by major public health events based on the sampling date. SBSs from each SARS-CoV-2 sequence were identified relative to the MN996528.1 reference genome. Catalogues of SBS types and counts were generated to estimate the impact of selection in each open reading frame, and identify mutation clusters. The estimation of mutational fitness over time was performed using the Augur pipeline.</p><p><strong>Results: </strong>The biases in SBS types and proportions observed support previous reports of host antiviral defense activity involving the SARS-CoV-2 genome. There was an increase in U>C substitutions associated with adenosine deaminase acting on RNA (ADAR) activity uniquely observed during Epoch 4. The burden of novel SBSs observed in SARS-CoV-2 genomic sequences was the greatest in Epoch 2 (median 5), followed by Epoch 3 (median 4). Clusters of SBSs were observed in the spike protein open reading frame, ORF1a, and ORF3a. The high proportion of nonsynonymous SBSs and increasing dN/dS metric (ratio of nonsynonymous to synonymous mutations in a given open reading frame) to above 1 in Epoch 4 indicate positive selection of the spike protein open reading frame.</p><p><strong>Conclusions: </strong>Quantitative analysis of the mutational patterns of the SARS-CoV-2 genome in the microcosm of Ontario, Canada within early consecutive epochs of the pandemic tracked the mutational dynamics in the context of public health events that instigate significant shifts in selection and mutagenesis. Continued genomic surveillance of emergent variants will be useful for the design of public health policies in response to the evolving COVID-19 pandemic.</p>","PeriodicalId":73552,"journal":{"name":"JMIR bioinformatics and biotechnology","volume":" ","pages":"e42243"},"PeriodicalIF":0.0000,"publicationDate":"2022-12-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11135226/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JMIR bioinformatics and biotechnology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2196/42243","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background: The emergence of SARS-CoV-2 variants with mutations associated with increased transmissibility and virulence is a public health concern in Ontario, Canada. Characterizing how the mutational patterns of the SARS-CoV-2 genome have changed over time can shed light on the driving factors, including selection for increased fitness and host immune response, that may contribute to the emergence of novel variants. Moreover, the study of SARS-CoV-2 in the microcosm of Ontario, Canada can reveal how different province-specific public health policies over time may be associated with observed mutational patterns as a model system.
Objective: This study aimed to perform a comprehensive analysis of single base substitution (SBS) types, counts, and genomic locations observed in SARS-CoV-2 genomic sequences sampled in Ontario, Canada. Comparisons of mutational patterns were conducted between sequences sampled during 4 different epochs delimited by major public health events to track the evolution of the SARS-CoV-2 mutational landscape over 2 years.
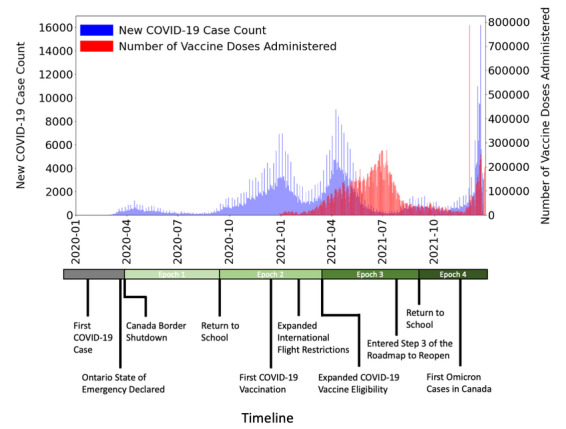
Methods: In total, 24,244 SARS-CoV-2 genomic sequences and associated metadata sampled in Ontario, Canada from January 1, 2020, to December 31, 2021, were retrieved from the Global Initiative on Sharing All Influenza Data database. Sequences were assigned to 4 epochs delimited by major public health events based on the sampling date. SBSs from each SARS-CoV-2 sequence were identified relative to the MN996528.1 reference genome. Catalogues of SBS types and counts were generated to estimate the impact of selection in each open reading frame, and identify mutation clusters. The estimation of mutational fitness over time was performed using the Augur pipeline.
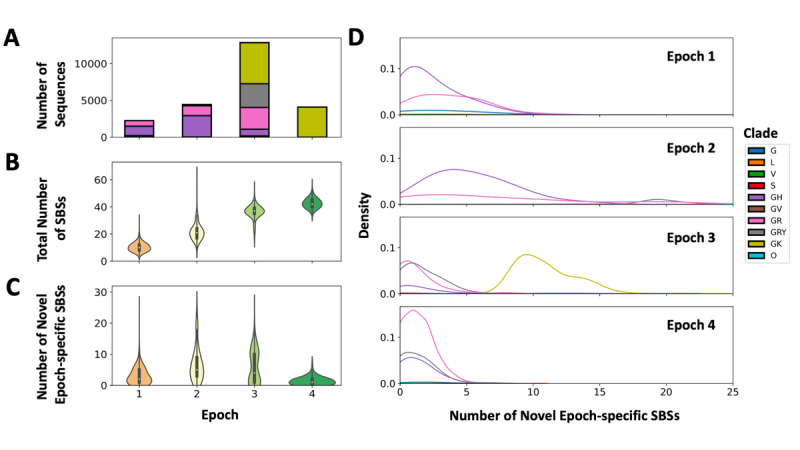
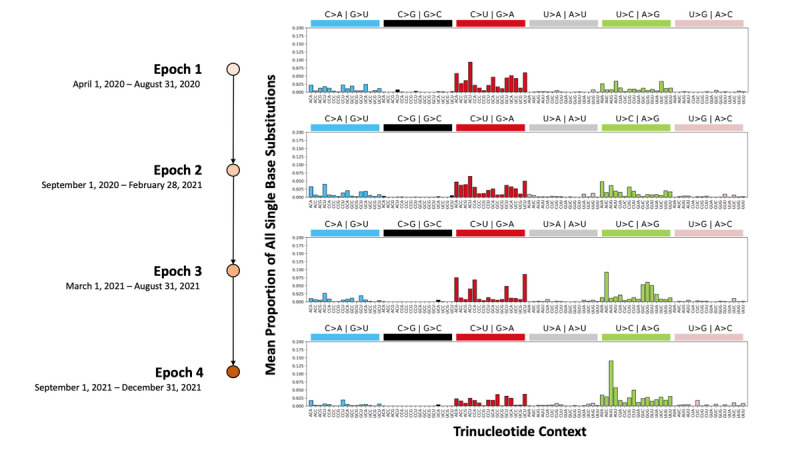
Results: The biases in SBS types and proportions observed support previous reports of host antiviral defense activity involving the SARS-CoV-2 genome. There was an increase in U>C substitutions associated with adenosine deaminase acting on RNA (ADAR) activity uniquely observed during Epoch 4. The burden of novel SBSs observed in SARS-CoV-2 genomic sequences was the greatest in Epoch 2 (median 5), followed by Epoch 3 (median 4). Clusters of SBSs were observed in the spike protein open reading frame, ORF1a, and ORF3a. The high proportion of nonsynonymous SBSs and increasing dN/dS metric (ratio of nonsynonymous to synonymous mutations in a given open reading frame) to above 1 in Epoch 4 indicate positive selection of the spike protein open reading frame.
Conclusions: Quantitative analysis of the mutational patterns of the SARS-CoV-2 genome in the microcosm of Ontario, Canada within early consecutive epochs of the pandemic tracked the mutational dynamics in the context of public health events that instigate significant shifts in selection and mutagenesis. Continued genomic surveillance of emergent variants will be useful for the design of public health policies in response to the evolving COVID-19 pandemic.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
