Simon Houston, Karen Vivien Lithgow, Kara Krista Osbak, Chris Richard Kenyon, Caroline E. Cameron
{"title":"Functional insights from proteome-wide structural modeling of Treponema pallidum subspecies pallidum, the causative agent of syphilis","authors":"Simon Houston, Karen Vivien Lithgow, Kara Krista Osbak, Chris Richard Kenyon, Caroline E. Cameron","doi":"10.1186/s12900-018-0086-3","DOIUrl":null,"url":null,"abstract":"<p>Syphilis continues to be a major global health threat with 11 million new infections each year, and a global burden of 36 million cases. The causative agent of syphilis, <i>Treponema pallidum</i> subspecies <i>pallidum</i>, is a highly virulent bacterium, however the molecular mechanisms underlying <i>T. pallidum</i> pathogenesis remain to be definitively identified. This is due to the fact that <i>T. pallidum</i> is currently uncultivatable, inherently fragile and thus difficult to work with, and phylogenetically distinct with no conventional virulence factor homologs found in other pathogens. In fact, approximately 30% of its predicted protein-coding genes have no known orthologs or assigned functions. Here we employed a structural bioinformatics approach using Phyre2-based tertiary structure modeling to improve our understanding of <i>T. pallidum</i> protein function on a proteome-wide scale.</p><p>Phyre2-based tertiary structure modeling generated high-confidence predictions for 80% of the <i>T. pallidum</i> proteome (780/978 predicted proteins). Tertiary structure modeling also inferred the same function as primary structure-based annotations from genome sequencing pipelines for 525/605 proteins (87%), which represents 54% (525/978) of all <i>T. pallidum</i> proteins. Of the 175?<i>T. pallidum</i> proteins modeled with high confidence that were not assigned functions in the previously annotated published proteome, 167 (95%) were able to be assigned predicted functions. Twenty-one of the 175 hypothetical proteins modeled with high confidence were also predicted to exhibit significant structural similarity with proteins experimentally confirmed to be required for virulence in other pathogens.</p><p>Phyre2-based structural modeling is a powerful bioinformatics tool that has provided insight into the potential structure and function of the majority of <i>T. pallidum</i> proteins and helped validate the primary structure-based annotation of more than 50% of all <i>T. pallidum</i> proteins with high confidence. This work represents the first <i>T. pallidum</i> proteome-wide structural modeling study and is one of few studies to apply this approach for the functional annotation of a whole proteome.</p>","PeriodicalId":51240,"journal":{"name":"BMC Structural Biology","volume":"18 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2018-05-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12900-018-0086-3","citationCount":"14","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/s12900-018-0086-3","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 14
Abstract
Syphilis continues to be a major global health threat with 11 million new infections each year, and a global burden of 36 million cases. The causative agent of syphilis, Treponema pallidum subspecies pallidum, is a highly virulent bacterium, however the molecular mechanisms underlying T. pallidum pathogenesis remain to be definitively identified. This is due to the fact that T. pallidum is currently uncultivatable, inherently fragile and thus difficult to work with, and phylogenetically distinct with no conventional virulence factor homologs found in other pathogens. In fact, approximately 30% of its predicted protein-coding genes have no known orthologs or assigned functions. Here we employed a structural bioinformatics approach using Phyre2-based tertiary structure modeling to improve our understanding of T. pallidum protein function on a proteome-wide scale.
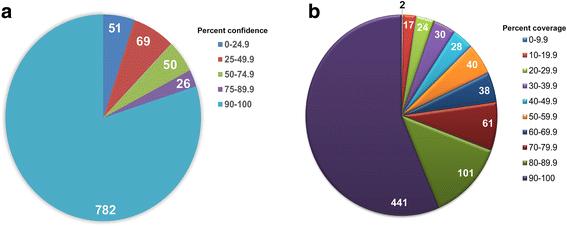
Phyre2-based tertiary structure modeling generated high-confidence predictions for 80% of the T. pallidum proteome (780/978 predicted proteins). Tertiary structure modeling also inferred the same function as primary structure-based annotations from genome sequencing pipelines for 525/605 proteins (87%), which represents 54% (525/978) of all T. pallidum proteins. Of the 175?T. pallidum proteins modeled with high confidence that were not assigned functions in the previously annotated published proteome, 167 (95%) were able to be assigned predicted functions. Twenty-one of the 175 hypothetical proteins modeled with high confidence were also predicted to exhibit significant structural similarity with proteins experimentally confirmed to be required for virulence in other pathogens.
Phyre2-based structural modeling is a powerful bioinformatics tool that has provided insight into the potential structure and function of the majority of T. pallidum proteins and helped validate the primary structure-based annotation of more than 50% of all T. pallidum proteins with high confidence. This work represents the first T. pallidum proteome-wide structural modeling study and is one of few studies to apply this approach for the functional annotation of a whole proteome.
期刊介绍:
BMC Structural Biology is an open access, peer-reviewed journal that considers articles on investigations into the structure of biological macromolecules, including solving structures, structural and functional analyses, and computational modeling.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
