{"title":"Destabilization of the TWIST1/E12 complex dimerization following the R154P point-mutation of TWIST1: an in silico approach","authors":"Charlotte Bouard, Raphael Terreux, Agnès Tissier, Laurent Jacqueroud, Arnaud Vigneron, Stéphane Ansieau, Alain Puisieux, Léa Payen","doi":"10.1186/s12900-017-0076-x","DOIUrl":null,"url":null,"abstract":"<p>The bHLH transcription factor TWIST1 plays a key role in the embryonic development and in tumorigenesis. Some loss-of-function mutations of the <i>TWIST1</i> gene have been shown to cause an autosomal dominant craniosynostosis, known as the Saethre-Chotzen syndrome (SCS). Although the functional impacts of many <i>TWIST1</i> mutations have been experimentally reported, little is known on the molecular mechanisms underlying their loss-of-function. In a previous study, we highlighted the predictive value of <i>in silico</i> molecular dynamics (MD) simulations in deciphering the molecular function of TWIST1 residues.</p><p>Here, since the substitution of the arginine 154 amino acid by a glycine residue (R154G) is responsible for the SCS phenotype and the substitution of arginine 154 by a proline experimentally decreases the dimerizing ability of TWIST1, we investigated the molecular impact of this point mutation using MD approaches. Consistently, MD simulations highlighted a clear decrease in the stability of the α-helix during the dimerization of the mutated R154P TWIST1/E12 dimer compared to the wild-type TE complex, which was further confirmed in vitro using immunoassays.</p><p>Our study demonstrates that MD simulations provide a structural explanation for the loss-of-function associated with the SCS TWIST1 mutation and provides a proof of concept of the predictive value of these MD simulations. This <i>in silico</i> methodology could be used to determine reliable pharmacophore sites, leading to the application of docking approaches in order to identify specific inhibitors of TWIST1 complexes.</p>","PeriodicalId":498,"journal":{"name":"BMC Structural Biology","volume":"17 1","pages":""},"PeriodicalIF":2.2220,"publicationDate":"2017-05-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12900-017-0076-x","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/s12900-017-0076-x","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 4
Abstract
The bHLH transcription factor TWIST1 plays a key role in the embryonic development and in tumorigenesis. Some loss-of-function mutations of the TWIST1 gene have been shown to cause an autosomal dominant craniosynostosis, known as the Saethre-Chotzen syndrome (SCS). Although the functional impacts of many TWIST1 mutations have been experimentally reported, little is known on the molecular mechanisms underlying their loss-of-function. In a previous study, we highlighted the predictive value of in silico molecular dynamics (MD) simulations in deciphering the molecular function of TWIST1 residues.
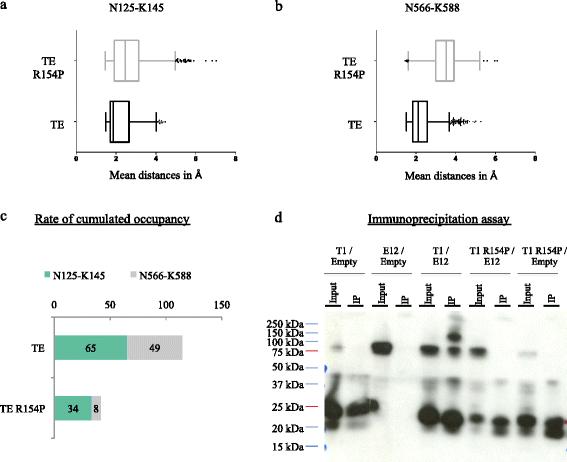
Here, since the substitution of the arginine 154 amino acid by a glycine residue (R154G) is responsible for the SCS phenotype and the substitution of arginine 154 by a proline experimentally decreases the dimerizing ability of TWIST1, we investigated the molecular impact of this point mutation using MD approaches. Consistently, MD simulations highlighted a clear decrease in the stability of the α-helix during the dimerization of the mutated R154P TWIST1/E12 dimer compared to the wild-type TE complex, which was further confirmed in vitro using immunoassays.
Our study demonstrates that MD simulations provide a structural explanation for the loss-of-function associated with the SCS TWIST1 mutation and provides a proof of concept of the predictive value of these MD simulations. This in silico methodology could be used to determine reliable pharmacophore sites, leading to the application of docking approaches in order to identify specific inhibitors of TWIST1 complexes.
期刊介绍:
BMC Structural Biology is an open access, peer-reviewed journal that considers articles on investigations into the structure of biological macromolecules, including solving structures, structural and functional analyses, and computational modeling.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
