{"title":"A structural model of the E. coli PhoB Dimer in the transcription initiation complex","authors":"Chang-Shung Tung, Benjamin H McMahon","doi":"10.1186/1472-6807-12-3","DOIUrl":null,"url":null,"abstract":"<p>There exist > 78,000 proteins and/or nucleic acids structures that were determined experimentally. Only a small portion of these structures corresponds to those of protein complexes. While homology modeling is able to exploit knowledge-based potentials of side-chain rotomers and backbone motifs to infer structures for new proteins, no such general method exists to extend our understanding of protein interaction motifs to novel protein complexes.</p><p>We use a Motif Binding Geometries (MBG) approach, to infer the structure of a protein complex from the database of complexes of homologous proteins taken from other contexts (such as the helix-turn-helix motif binding double stranded DNA), and demonstrate its utility on one of the more important regulatory complexes in biology, that of the RNA polymerase initiating transcription under conditions of phosphate starvation. The modeled PhoB/RNAP/σ-factor/DNA complex is stereo-chemically reasonable, has sufficient interfacial Solvent Excluded Surface Areas (SESAs) to provide adequate binding strength, is physically meaningful for transcription regulation, and is consistent with a variety of known experimental constraints.</p><p>Based on a straightforward and easy to comprehend concept, \"proteins and protein domains that fold similarly could interact similarly\", a structural model of the PhoB dimer in the transcription initiation complex has been developed. This approach could be extended to enable structural modeling and prediction of other bio-molecular complexes. Just as models of individual proteins provide insight into molecular recognition, catalytic mechanism, and substrate specificity, models of protein complexes will provide understanding into the combinatorial rules of cellular regulation and signaling.</p>","PeriodicalId":498,"journal":{"name":"BMC Structural Biology","volume":"12 1","pages":""},"PeriodicalIF":2.2220,"publicationDate":"2012-03-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1472-6807-12-3","citationCount":"7","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/1472-6807-12-3","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 7
Abstract
There exist > 78,000 proteins and/or nucleic acids structures that were determined experimentally. Only a small portion of these structures corresponds to those of protein complexes. While homology modeling is able to exploit knowledge-based potentials of side-chain rotomers and backbone motifs to infer structures for new proteins, no such general method exists to extend our understanding of protein interaction motifs to novel protein complexes.
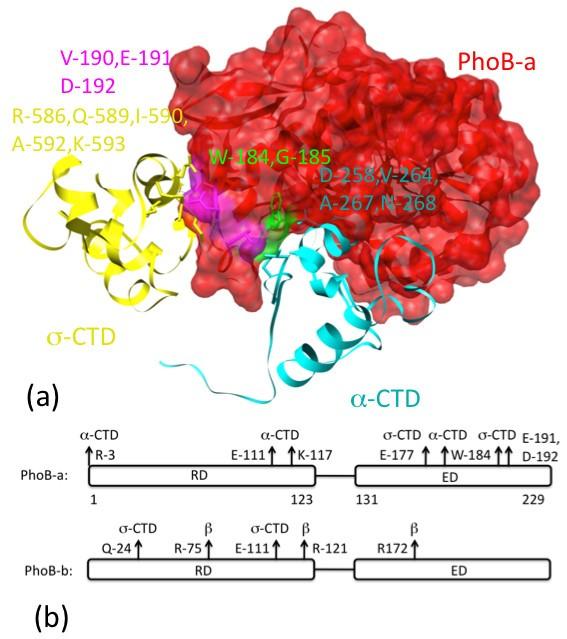
We use a Motif Binding Geometries (MBG) approach, to infer the structure of a protein complex from the database of complexes of homologous proteins taken from other contexts (such as the helix-turn-helix motif binding double stranded DNA), and demonstrate its utility on one of the more important regulatory complexes in biology, that of the RNA polymerase initiating transcription under conditions of phosphate starvation. The modeled PhoB/RNAP/σ-factor/DNA complex is stereo-chemically reasonable, has sufficient interfacial Solvent Excluded Surface Areas (SESAs) to provide adequate binding strength, is physically meaningful for transcription regulation, and is consistent with a variety of known experimental constraints.
Based on a straightforward and easy to comprehend concept, "proteins and protein domains that fold similarly could interact similarly", a structural model of the PhoB dimer in the transcription initiation complex has been developed. This approach could be extended to enable structural modeling and prediction of other bio-molecular complexes. Just as models of individual proteins provide insight into molecular recognition, catalytic mechanism, and substrate specificity, models of protein complexes will provide understanding into the combinatorial rules of cellular regulation and signaling.
期刊介绍:
BMC Structural Biology is an open access, peer-reviewed journal that considers articles on investigations into the structure of biological macromolecules, including solving structures, structural and functional analyses, and computational modeling.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
