{"title":"The origin of β-strand bending in globular proteins","authors":"Kazuo Fujiwara, Shinichi Ebisawa, Yuka Watanabe, Hiromi Fujiwara, Masamichi Ikeguchi","doi":"10.1186/s12900-015-0048-y","DOIUrl":null,"url":null,"abstract":"<p>Many β-strands are not flat but bend and/or twist. However, although almost all β-strands have a twist, not all have a bend, suggesting that the underlying force(s) driving β-strand bending is distinct from that for the twist. We, therefore, investigated the physical origin(s) of β-strand bends.</p><p>We calculated rotation, twist and bend angles for a four-residue short frame. Fixed-length fragments consisting of six residues found in three consecutive short frames were used to evaluate the twist and bend angles of full-length β-strands.</p><p>We calculated and statistically analyzed the twist and bend angles of β-strands found in globular proteins with known three-dimensional structures. The results show that full-length β-strand bend angles are related to the nearby aromatic residue content, whereas local bend angles are related to the nearby aliphatic residue content. Furthermore, it appears that β-strands bend to maximize their hydrophobic contacts with an abutting hydrophobic surface or to form a hydrophobic side-chain cluster when an abutting hydrophobic surface is absent.</p><p>We conclude that the dominant driving force for full-length β-strand bends is the hydrophobic interaction involving aromatic residues, whereas that for local β-strand bends is the hydrophobic interaction involving aliphatic residues.</p>","PeriodicalId":51240,"journal":{"name":"BMC Structural Biology","volume":"15 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2015-10-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12900-015-0048-y","citationCount":"5","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/s12900-015-0048-y","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 5
Abstract
Many β-strands are not flat but bend and/or twist. However, although almost all β-strands have a twist, not all have a bend, suggesting that the underlying force(s) driving β-strand bending is distinct from that for the twist. We, therefore, investigated the physical origin(s) of β-strand bends.
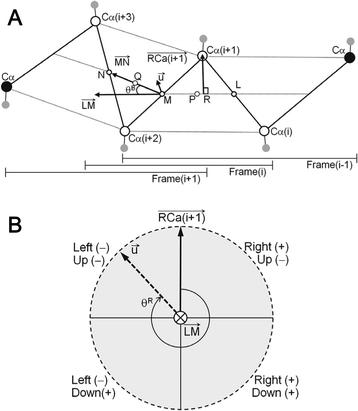
We calculated rotation, twist and bend angles for a four-residue short frame. Fixed-length fragments consisting of six residues found in three consecutive short frames were used to evaluate the twist and bend angles of full-length β-strands.
We calculated and statistically analyzed the twist and bend angles of β-strands found in globular proteins with known three-dimensional structures. The results show that full-length β-strand bend angles are related to the nearby aromatic residue content, whereas local bend angles are related to the nearby aliphatic residue content. Furthermore, it appears that β-strands bend to maximize their hydrophobic contacts with an abutting hydrophobic surface or to form a hydrophobic side-chain cluster when an abutting hydrophobic surface is absent.
We conclude that the dominant driving force for full-length β-strand bends is the hydrophobic interaction involving aromatic residues, whereas that for local β-strand bends is the hydrophobic interaction involving aliphatic residues.
期刊介绍:
BMC Structural Biology is an open access, peer-reviewed journal that considers articles on investigations into the structure of biological macromolecules, including solving structures, structural and functional analyses, and computational modeling.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
